
Part:BBa_K1319004
TEV protease with anti-self cleavage mutation S219V, codon optimized for E. coli
Gold criteria iGEM19_Montpellier
iGEM19_Montpellier offers an improved version of this TEV by adding a VHH. Montpellier characterize a more important enzyme activity against its substrate through the addition of a VHH that helps it to find its target. Please do not hesitate to consult the main page of this new parts.
iGEM19_Montpellier reports an improvement of this TEV: (Part:BBa_K3147009)by adding a VHH to it
Resume
This part is a TEV protease in RFC25 that was optimized for expression in E. coli. The part contains the S219V anti-self cleavage mutation.
The TEV Protease (also known as Tobaco Edge Virus nuclear inclusion a endopeptidase) is a highly sequence specific cysteine protease from the Tobacco Edge Virus (TEV). The protease is highly sequence specific. The consensus sequence for the cut is ENLYFQ\S with \ denoting the cleaved peptide bond. This sequence can be found in the part K1319016.
ENLYFQ\S is the optimal cleavage site with the highest activity but the protease is also active to a greater or lesser extent on a range of substrates. The highest cleavage is of sequences closest to the consensus EXLYΦQ\φ where X is any residue, Φ is any large or medium hydrophobic amino acid and φ is any small hydrophobic amino acid.
The TEV protease is commonly used as a biochemical tool to cleave affinity tags from purified proteins like His-Tags. The high specifity makes the protease relatively non-toxic in vitro and in vivo. The molecular weight of the TEV protease is 27 kDa.
Usage and Biology
The TEV Protease was used and characterizes in the K1319008 construct.
To characterize the TEV protease we used the fusion protein K1319014. This fusion protein contains GFP (E0040) bound to a dark quencher (REACh2/K1319002) over a linker which includes the TEV protease cleavage site. If the TEV protease successfully cuts the linker, GFP and its quencher would separate and the FRET (Förster Resonance Energy Transfer) system would be shut down. This would result in an increased GFP fluorescence.
To demonstrate this behaviour a double plasmid system was designed using the biobrick K1319013 in a pSB3K3 backbone and K1319008 in a pSB1C3 backbone. Also I20260 was used as a positive control because it produces the same GFP as used in the fusion protein and is regulated by the same promoter, RBS and Terminator on the same plasmid backbone. B0015 was used as negative control. Induction of the double plasmid constructs occured after 2 h with 50 µl of 100mM IPTG in a 50 ml shake flask culture.

|
| Comparison of the fluorescence adjusted for OD of I20260, B0015 and the double plasmid system K1319014 + K1319008 After induction with IPTG after 2 h the double plasmid system produced a fast fluorescence response with an over 10-fold increase compared to the non induced state. I20260 served as positive control and B0015 as negative control. |
The increase in fluorescence after induction with IPTG is clear sign of funtional expression of the TEV protease. The difference between not induced and induced plasmid is proof that the increase in fluorescence is only attributed to the successful cleavage of the linker. Therefore this is proof of a functional expression of the TEV protease after induction with IPTG.
Furthermore we validated the used plasmid with a Check PCR with one primer binding upstream on the plasmid backbone and one specifically in the insert.

|
| Check PCR for K1319008 The length of the PCR product matches the length of the control plasmid. |
This validates that the construct is indeed the TEV protease and thereby the functionality of the TEV protease is established. The construct K1319008 was also sequenced. The sequencing data can be seen in the parts registry here.
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
Characterization-CSMU_Taiwan2019
This part was characterized by CSMU_Taiwan 2019.Because of its stringent sequence specificity, tobacco etch virus (TEV) protease emerges as a useful reagent with wide application in the cleavage of recombinant fusion proteins. Thus, we have chosen TEV protease(sequence from BBa_K1319004) to remove the fusion protein in our another part BBa_K2951008 and be characterized.
Cloning
After obtaining TEV protease sequence in pSB1C3 from iGEM 2019 distribution kit(plate 6 21N), transforming it into DH5α and performing plasmid purification, we obtained the amount of plasmids required for construction. To characterize the TEV protease, the chosen vector for expression is pET29a which includes a 6xHis-Tag fusion on the plasmid following the insert. At both anchors at the N- and C-terminus the restriction sites for BamHI and XhoI were introduced to clone the genes into a pET29a vector by PCR with overhang primers primer1(TEV-fwd) and primer2(TEV-rev). The PCR cycle number was 40 repeating step 2-4, the detailed conditions are shown in the table below.
| Name | 5'-3' primers sequences |
|---|---|
| TEV-fwd | AATTGCGGATCCGGCGAAAGCCTGTTCAAAGGTC |
| TEV-rev | AATTGCCTCGAGACCACCTTGGGAGTAGACCAGTTCATTC |
| Step | Condition |
|---|---|
| 1 Plasmid Denature | 94°C,5' |
| 2 dsDNA Denature | 94°C,20 |
| 3 Annealing | 55°C,20 |
| 4 Extension | 72°C,35 |
| 5 Final Extension | 72°C,2' |
| 6 Hold | 4°C |
The PCR result is observed by running a 1.8% gel and further purified(Fig.1). The modified TEV insertion were first digested with the restriction enzymes(BamH1 and Xho1)and so did the pET29a vector. (Fig.2) (We digested the insertion and vector one by one because the amount of DNA decreased significantly after the digestions on our first double digestion, and we assumed that this is the result of star effect.)
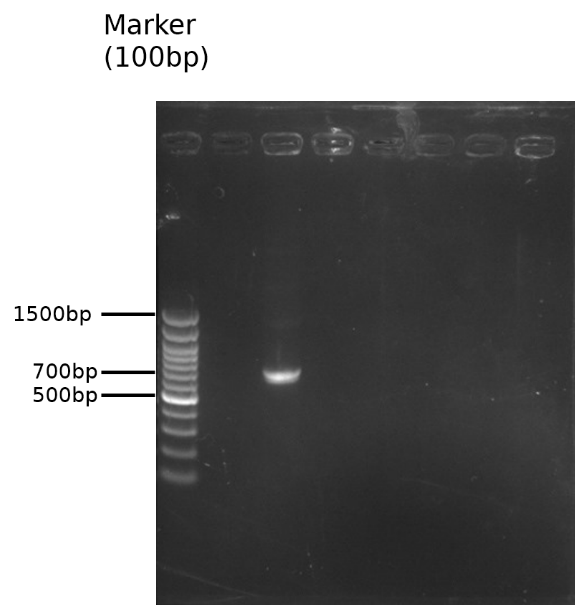
Fig.1 1.8% gel of pSB1C3-BBa_K1319004 overhang PCR and DNA purification

Fig.2 0.8% gel of pET29a digestion(the 3kb is from the original insert of the vector)
After digestion, we ligated the plasmid and the TEV insert using the T4 ligase. This sample was then transformed in E.coli DH5α for colony PCR(using primer-T7 promoter and terminator) to confirm that we have successfully inserted the TEV protease sequence into pET29a(Fig.3).The sequence of two colony with the cloned part was confirmed from sequencing results. All the bases of the cloned part were confirmed to be correct.
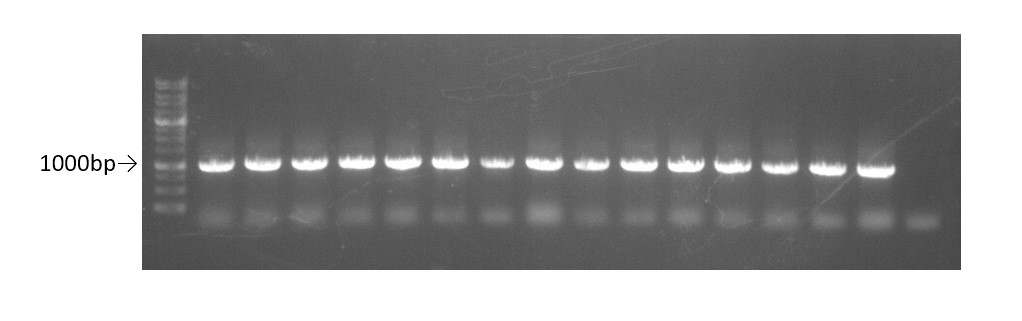
Fig.3 0.8% gel of colony PCR using primers T7 promoter and terminator for pET29a.
Well 1-15:pET29a-TEV insert plasmid.
Well 16: control-pET29a with original insertion.
Small scale production
Cultivations and Induction of protein expression
For small scale production, the plasmid was transformed into E.coli BL21. 1 colony from the transformation plates were inoculated in 10 ml LB-kanamycin (50 μg/ml working concentration) and grew the cells at +37 °C, 150rpm for 12-18hrs. 1cc was taken out to centrifuge and added 20ml of LB-kanamycin (50 μg/ml) to make the OD value~0.1. After incubation for another 2 hrs, it reached the OD595 value 0.4~0.7. (if the OD value exceeds 0.7, the culture would be diluted again.) When finished growing the cells, the 20ml was split into two, and one of them was induced to express the gene of interest by adding a final concentration of 0,5 mM IPTG in the cultures and the other tube as the uninduced control. Both were shaked at +37 °C for 2.5hrs. We then tested their OD value and take V ml(V=2/OD) from each tube for centrifugation at 12000rpm. The induction result was checked by SDS-PAGE coomassie blue staining (Fig.4) Good expression can be clearly seen in both lane 3(without induction by adding IPTG) and 4, indicating obvious leakage.
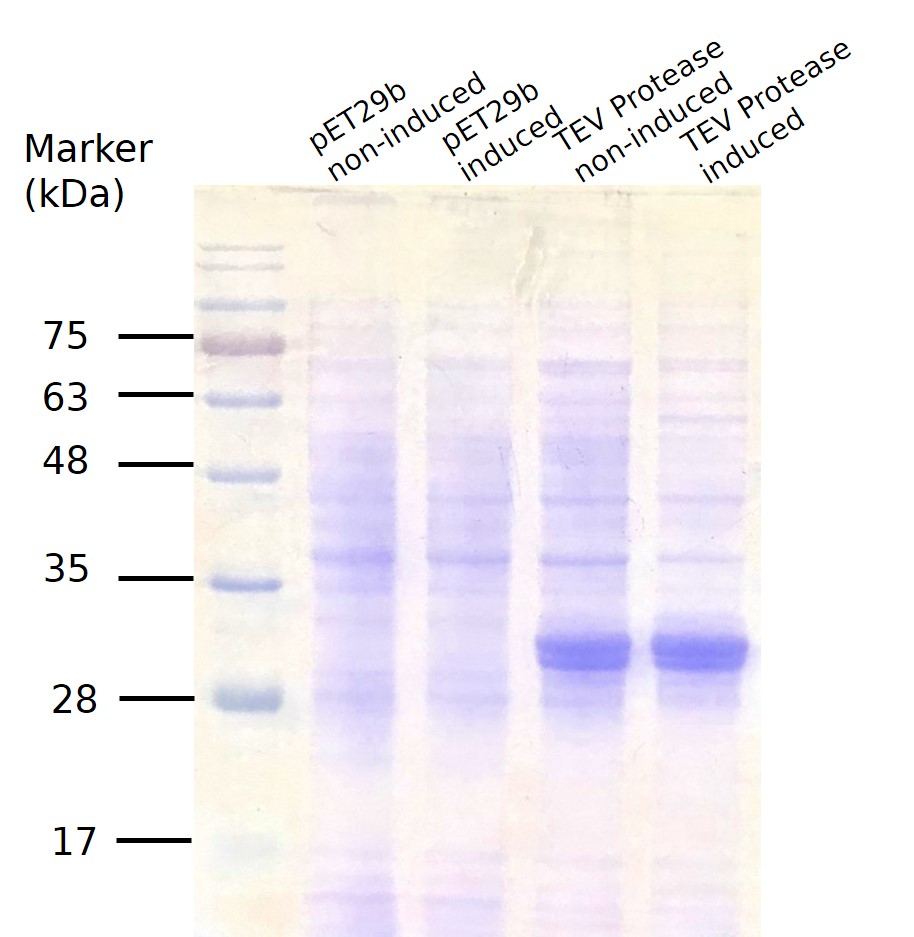
Fig.4 SDS PAGE for TEV Protease small scale production.
Large scale production
Cultivations and Induction of protein expression
2 colonies from the transformation plates were inoculated in two tubes of 15 ml LB-kanamycin (50 μg/ml working concentration) and grew the cells at +37 °C, 150rpm for 12-18hrs. 8cc was taken out from each tube to centrifuge and diluted with LB-kanamycin (50 μg/ml) to 200ml to make the OD value~0.1. After incubation for another 2 hrs, it reached the OD595 value 0.4~0.7. (if the OD value exceeds 0.7, the culture would be diluted again.) IPTG was further added and the following incubation was 2 hrs.
Protein solubility analysis
To further characterize the solubility of this part, we then sonicated the culture and did 8700G and 16,000G centrifugation. We then did SDS-PAGE for coomassie brilliant blue staining and western blot to detect the content of our protein(Fig.5). In Fig.5, there was more “8700G P” group when compared with the “8700G S” group. This result meant that most proteins were deposited in the cell pellet after 8700G centrifugation because they are expressed as insoluble forms. Thus, minimal amount of TEV protease could be extracted from the cell lysate.

Fig.5 T meant the initial sample obtained after sonication; 8700G P and 8700G S meant the pellet and the supernatant obtained after 8700G for 20 min; 8700G S and 16,000G T meant the supernatant gotten after 8700G for 20 min;16,000G P and 16,000G S meant the pellet and the supernatant obtained after 16,000G for 20 min.
Purification and Dialysis
After extracting the cell lysates, we used nickel-resin column to purify our target proteins from the cell lysates because all of our proteins were tagged with 6 histidines fusion at their C-terminal ends with the pET29a plasmid. After protein purification, protein dialysis with PBS buffer to remove imidazole in our purified proteins, we did SDS-PAGE gel electrophoresis to ensure our target proteins were purified. However, after various troubleshooting, the results still shown poor yield of TEV protease most likely because of its insolubility. Expression with fusion proteins would be a possible way to obtain higher proportions.
Protein Expression overtime
We transformed the plasmids that contained this part into competent cell E.coli BL21. After cultured overnight, measure the ABS600 and diluting the LB medium to O.D.=0.1. Then incubated at 37℃, 150 rpm until the O.D. of the samples reached 0.4 to 0.6 . Add IPTG( final concentration : 0.4mM ) to 125 ml flask and another without adding IPTG as control, and then return to 37°C. From then on, after measure the O.D. values, transfer 1 ml from the induced sample and centrifuge at maximum speed for 60 seconds at RT and remove supernatant at 0, 0.5, 1.0, 1.5, 2.0 ,2.5 , 3.0, 3.5, 4 , 5, 6 hours. Then we use SDS PAGE to analyze the quantity of TEV protease at each time spot.
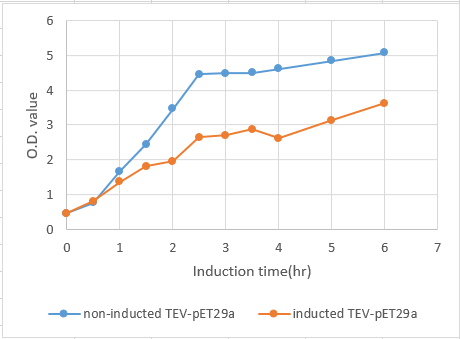
Fig.6 The growth curve of BL21 induced by IPTG from 0 to 6 hours. The concentration of BL21 reached stationary phase at 2.5 hours. Also, induction shows a significant effect on the growth rate of E.coli BL21 carrying TEV-pET29a.
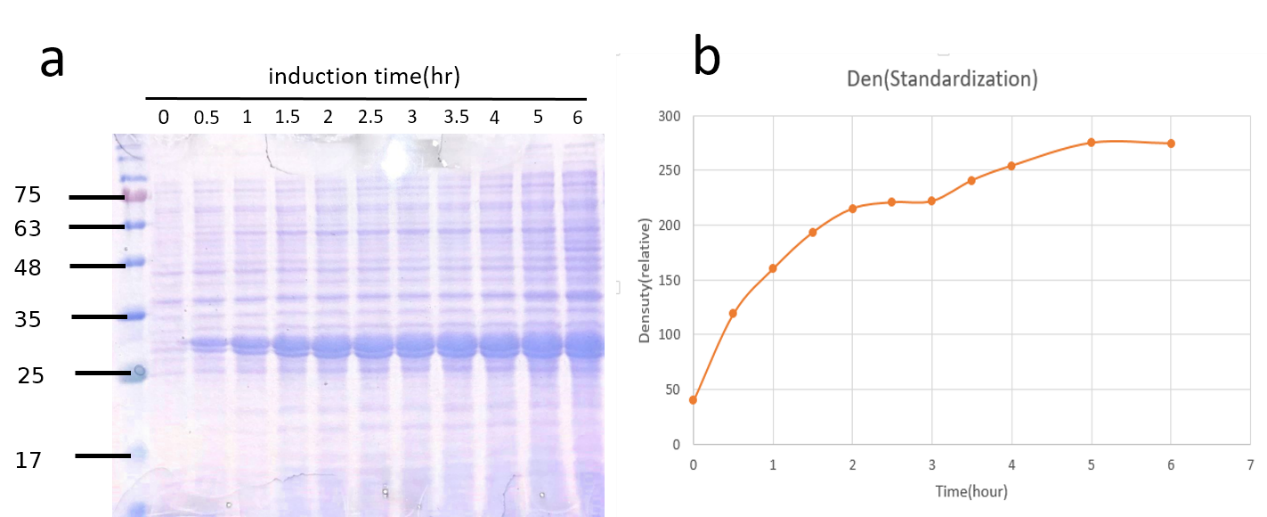
Fig.7a SDS-PAGE coomassie brilliant blue staining of TEV-pET29a induction. Fig.7b Quantification of TEV protease expression overtime.
Though the amount of TEV protease (28kDa) increased consistently with time, we could not jump to conclusions that it was proper to incubate E.coli as long as possible. Another consideration was the time it would take. Just as we expected, it growed fast at the first 2.5 hours. That’s why we also chose 2.5hr after induced by IPTG when we extracted it from total cell lysates.
| None |