
 1 Registry Star
1 Registry StarPart:BBa_I0500
Inducible pBad/araC promoter
pBad is an E. coli promoter that is tightly controlled by:
- inducer: L-[http://openwetware.org/wiki/Arabinose arabinose].
- repressor: AraC acts as the repressor
one [http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=pubmed&cmd=Retrieve&dopt=Abstract&list_uids=7768852 study] concluded that [http://openwetware.org/wiki/Arabinose arabinose] can change the conformation of araC and prevent it from successfully binding to and repressing pBad.
Usage and Biology
- When grown with 0.2% arabinose, promoter is weak-medium. [jb, 5/24/04] Part may not be compatible with MC4100 as cell line is araD 139.
- MC4100 is not a good chassis for operating BBa_I0500 (pBad promoter). The feed-forward regulation of the endogenous promoter controlling expression of the arabinose transporter prevents linear induction with increasing arabinose concentration. ((Engineered strain from Keasling's lab, used by jrk for operation of the screening plasmid.))
>Internal Priming Screening Characterization of BBa_I0500: Has 1 possible internal priming site between this BioBrick part and the VF2 primer.
The 2018 Hawaii iGEM team evaluated the 40 most frequently used BioBricks and ran them through an internal priming screening process that we developed using the BLAST program tool. Out of the 40 BioBricks we evaluated, 10 of them showed possible internal priming of either the VF2 or VR primers and sometime even both. The data set has a range of sequence lengths from as small as 12 bases to as large as 1,210 bases. We experienced the issue of possible internal priming during the sequence verification process of our own BBa_K2574001 BioBrick and in the cloning process to express the part as a fusion protein. BBa_K2574001 is a composite part containing a VLP forming Gag protein sequence attached to a frequently used RFP part (BBa_E1010). We conducted a PCR amplification of the Gag-RFP insert using the VF2 and VR primers on the ligation product (pSB1C3 ligated to the Gag + RFP). This amplicon would serve as template for another PCR where we would add the NcoI and BamHI restriction enzyme sites through new primers for ligation into pET14b and subsequent induced expression. Despite gel confirming a rather large, approximately 2.1 kb insert band, our sequencing results with the VR primer and BamHI RFP reverse primer gave mixed results. Both should have displayed the end of the RFP, but the VR primer revealed the end of the Gag. Analysis of the VR primer on the Gag-RFP sequence revealed several sites where the VR primer could have annealed with ~9 - 12 bp of complementarity. Internal priming of forward and reverse primers can be detrimental to an iGEM project because you can never be sure if the desired construct was correctly inserted into the BioBrick plasmid without a successful sequence verification.
For the BioBrick part BBa_I0500, the location of the internal priming site is on the 1151-1158 base number of the BioBrick and on the 5-12 base number of the VF2 primer.
[http://2010.igem.org/Team:Slovenia Team Slovenia 2010] further characterized pBAD promoter. Check results on Experience.
[http://2016.igem.org/Team:IISc_Bangalore IISc Bangalore 2016] showed catabolite repression of expression from the promoter by glucose. Check results on Experience.
[http://2016.igem.org/Team:OUC-China OUC-China 2016] characterized BBa_I0500 of different concentrations of L-arabinose on the transcriptional level. Check results on Experience.
[http://2017.igem.org/Team:TU-Eindhoven TU-Eindhoven 2017] optimized the expression on by testing 3 different temperatures, where 37 degrees Celcius proved most successful. Addition of L-arabinose has also been optimized to compensate for the breakdown of L-arabinose by the E.coli strain BL21(DE3) for mCherry and GFP expression. Check results on Experience.
[http://2017.igem.org/Team:Glasgow Team Glasgow 2017] improved this part by splitting its constituent parts into two separate BioBricks: BBa_K2442101 encoding minimal pBAD and BBa_K2442104 encoding AraC under regulation of LacI-regulated promoter. This allows for greater control over arabinose-inducible systems. See details on Experience.
[http://2019.igem.org/Team:BHSF_ND BHSF_ND 2019] Characterised the efficiency on the function of translating fluorescence under different inducer concentration at the given time of 240 minutes and under different time stage at a given inducer concentration.Also, we find out that at a certain time node that the system will suddenly increase its productivity. At last, the result suggested that both system have noticeable leakage and Xyls has much higher leakage. Check results on Experience.
Jilin_China 2020 This year, based on the previous part BBa_I0500, Jilin_China characterized this part again and added new documentation to it. See more details in Proof_Of_Concept in Jilin_China 2020
Leiden 2021 Calibrate the efficiency for the production of the fluorescent proteins mCherry in E. coli TOP10. The fluorescent intensity was compared to a internal reference of constitutive promoter BBa_J23100 Check results on Experience.
[http://2022.igem.org/Team:Jilin_China Jilin_China 2022] Based on the previous part BBa_I0500, Jilin_China optimized our experimental design and characterized this part again, gaining the characterization results that we believe to be more accurate. See more details in Contribution in Jilin_China 2022
- [http://openwetware.org/wiki/Titratable_control_of_pBAD_and_lac_promoters_in_individual_E._coli_cells#pBAD_promotersOpenWetWare From an OWW article on pBAD and lac promoters]:
- Import of arabinose into cells is mediated by the araE gene. Induction of the arabinose transporter encoded by araE can be uncoupled from the endogenous PBAD promoter by deleting the chromosomal araE gene and replacing it with a plasmid-borne copy of araE under control of a constitutive promoter (1). However, this does not seem to be enough to allow for homogenous expression from PBAD promoters in a population of cells (2).
- At low concentrations of arabinose, degradation of the sugar within cells also effects the homogeneity of expression from PBAD promoters (2). Arabinose degradation is mediated by the araBAD genes. Strains lacking functional araE, araFGH (another transporter), and araBAD can be made to be responsive to arabinose for PBAD promoter induction (2). This is achieved by introduction of a mutant lacY gene. LacY A177C allows for downhill transport of arabinose, as well as maltose, palatinose, sucrose, and cellobiose (3), but does not actively transport these sugars (4). Lactose import is not affected in this mutant. So, PBAD promoters in cells lacking endogeneous arabinose importers and containing LacY A177C are linearly responsible to arabinose at the individual cell level.
- By the way, AraC is the repressor of the PBAD promoter. It is encoded on the pBAD vector series and is still present in the above-described strains.
PC and AraC are located on the complementary strand, reading right to left as written.
- At least one registry stock contains a deletion of the C at base 1194. This is after the transcriptional start but before the translation start, so it may not be significant. Parts with this mutation have been qualitatively observed to function normally.
Induction and Subsequent Inhibition of the pBAD Promoter
(Characterized by SDU-Denmark 2017)
Expression by the pBAD promoter can be regulated tightly by induction and subsequent inhibition.
The pBAD promoter holds great potential to control the expression of genes that require tight regulation, as it is capable of both an induction and repression. The [http://2015.igem.org/Team:HKUST-Rice, HKUST-Rice iGEM team] from 2015 found that the pBAD promoter exhibits an almost all-or-none behaviour upon induction with arabinose when located on a high copy vector, but allows for gradual induction when cloned into a low copy vector. Thus, it was evident that this promoter on a high copy vector would be inappropriate for tightly regulated gene expression. Based on these findings, a low copy vector was used to investigate the ability to inhibit gene expression subsequent to induction of pBAD.
Gene expression was simulated by fluorescence microscopy using a pBAD-YFP reporter system, BBa_I6058.
For this purpose, an Olympus IX83 with a photometrics prime camera was used with an exposure time for YFP at 200 ms. Transformed E. coli MG1655 cells were cultured in M9 minimal medium supplemented with 0.2% glycerol and 30 µg/mL chloramphenicol, to avoid catabolite repression from glucose residues present in LB medium. Two cultures were incubated, of which one was induced with 0.2 % arabinose from the beginning. At OD600=0.1, designated time 0, the cultures were split in two and 0.2 % glucose was added to one of each pair. Samples were obtained at time 0, before division of the cultures, and at 30 min, 60 min, and 120 min.
The resulting images revealed, that the inducer arabinose was required to stimulate expression of YFP, and that the addition of the repressor glucose to a uninduced culture had no effect. Furthermore, it was evident that addition of arabinose induced expression of YFP, and that subsequent addition of glucose terminated the pBAD regulated gene expression, resulting in a reduced fluorescence level. 30 minutes after inhibition this reduction was already evident, and after 120 minutes the gene expression controlled by pBAD was even further decreased, as seen in Figure 1.

Figure 1. YFP fluorescence levels in E. coli MG1655 transformed with the pBAD-YFP reporter system on pSB3K3. Left: Cultures with the inducer arabinose added. Right: Cultures not induced with arabinose. Both cultures were split up at OD600=0.1, designated time 0, and the inhibitor glucose was added to one half of each culture. Images were obtained at 0, 30, 60, and 120 minutes.
This experiment made it clear, that gene expression controlled by the pBAD promoter is both inducible and repressible as required when cloned into the low copy vector pSB3K3.
Induction of the pBAD Promoter
Characterized by Peking 2019
Promoter pBAD is induced with inexpensive and non-toxic monosaccharide L-arabinose and in bacterial strains deficient in arabinose catabolism. This promoter is tightly regulated and can reach moderately high levels of downstream gene expression.In order to obtain a dynamic range and proper concentration to induce the expression of dCas9, we use the cytometer to characterize the pBAD/araC promoter on respectively medium copy number pSB3C5 and low copy number pSB4A5 in TOP10 and found the fluorescense is increased as the concentration of arabinose increases (Figure 1).After we changed the downstream gene "sfGFP" with "sfGFP fused dCas9" on pSB3C5, it was observed that the expression of dCas9 from pBAD promoter can be modulated over a wide range of arabinose concentrations (Figure 2).
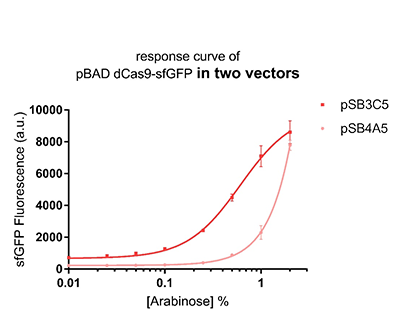
Figure 1: The response curve of pBAD dCas9-sfGFP in two vectors, respectively medium copy number pSB3C5 and low copy number pSB4A5
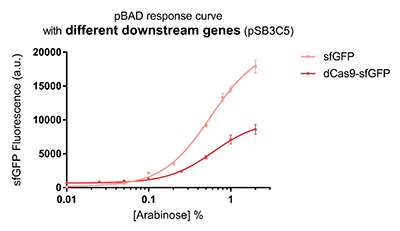
Figure 2: pBAD response curve with different downstream genes
The performance of two inducible promoters(AraC-pBAD compared to Xyls-Pm) in terms of time and concentration variation
Characterized by BHSF_ND 2019
The pBAD promoter holds great potential to control the expression of genes that require tight regulation, as it is capable of both an induction and repression. The pBAD promoter exhibits an almost all-or-none behavior upon induction with arabinose when located on a high copy vector, but allows for gradual induction when cloned into a low copy vector. Based on these research findings, a low copy vector was used to investigate the ability to inhibit gene expression subsequent to induction of pBAD.
As the result shows, the system exhibit relative high level of fluorescence compared to our backbone design( BN006/Contains BBa_K3202029) which suggest the inducible transcription system function properly.


Results
In our experiment of leakage testing, we used low-copying plasmid backbone PSB4K5, with a PSC101 origin of replication.
The inducer with their respective promoters (AraC & pBAD) are coupled with sfGFP to see if there is actually expression leakage when inducer is present quantitatively through flow cytometry. We tested fluorescence under different inducer concentration at the given time of 240 minutes and under different time stage at a given inducer concentration. Theoretically when the inducer (AraC) are present, the promoter (pBAD) is initiated therefore sfGFP is expressed; while sfGFP shouldn’t be expressed if inducer is absent. When measured at the end of 240 minutes without inducer, BN007/Contains BBa_K3202030(pBAD+sfGFP) has a basal leakage of 90 a.u. compared to BN006/Contains BBa_K3202029(backbone without GFP). The result suggested that this system have noticeable leakage.
When we gradually increased inducer concentration to 1mM, the fluorescence of BN006/Contains BBa_K3202029 stays at a constant level, whereas the fluorescence of pBAD have increased exponentially. This is another sign that the system function properly.
We found an intriguing result when measuring the fluorescence of the two system under a constant inducer concentration of 1mM. During the first 160 minutes, the fluorescence of BN007/Contains BBa_K3202030 is 200 a.u. higher than BN006/Contains BBa_K3202029. However, at 240 minutes, the fluorescence value suddenly increased to 7641 a.u., which is 7590 a.u., higher than the backbone. Therefore, in the subsequent experiments, systems are induced to up to 4 hours to ensure the accuracy of the experiment. Results shown above verified the presence of expression leakage of the system when inducer is not expressed through the detectable fluorescence of reporter protein using flow cytometry.
Functional Parameters: Austin_UTexas
Burden Imposed by this Part:
Burden is the percent reduction in the growth rate of E. coli cells transformed with a plasmid containing this BioBrick (± values are 95% confidence limits). This BioBrick did not exhibit a burden that was significantly greater than zero (i.e., it appears to have little to no impact on growth). Therefore, users can depend on this part to remain stable for many bacterial cell divisions and in large culture volumes. Refer to any one of the BBa_K3174002 - BBa_K3174007 pages for more information on the methods, an explanation of the sources of burden, and other conclusions from a large-scale measurement project conducted by the 2019 Austin_UTexas team.
This functional parameter was added by the 2020 Austin_UTexas team.
iGEM 2020 QHFZ-China, new documentation (For Bronze)
Group: QHFZ-China iGEM 2020
Author: Yixian Yang
TDPs (Tardigrade intrinsically Disordered Proteins) show a protective effect on bacteria. However, related studies are all used by the T7 promoter and E. coli BL21 (DE3) strain. This year, we used the Inducible pBad/araC promoter to express a TDP, CAHS 106094, in another strain. For all the experiments below, we use E. coli DH5α strain.
Part 1: Measurement with a reporter, GFP
Description
First, we measured the function of the promoter by GFP. We got the gene part from team NEFU-China 2020 as a gift.
Protocol
The gene circuit we used is as below:

The protocol is as below:
(1)Pick clones in good condition and put them into 500 μL LB medium containing antibiotics. Shake them to grow at 37℃ for 5~7 hours until the bacteria solution becomes turbid.
(2) Add 2mM iPTG into 3 mL LB medium containing antibiotics. Add 3 μL of the bacteria solution mentioned in step 1 to dilute the bacteria by the ratio of 1:1000. Shake the solution to grow the bacteria at 37℃ overnight.
(3) The bacteria solution was centrifuged, and the LB medium was removed. Then the bacteria were resuspended by PBS. 100 μL such solution was put into a well of a 96-well plate. The GFP fluorescence and OD600 were detected by microplate readers (Bio-Teck). The parameters are exciting light: 488 nm, light reception: 520 nm, gain 50.
(4) The value of PBS was deducted from the result above. GFP / OD600 was calculated.
Note: For step (3), The inducer usually was 0.2% L-arabinose. However, it can be changed if we want to study the concentration of the inducer and the effect of D-arabinose. To study whether glucose and trehalose affect the promoter, we even added D-glucose or D-trehalose of different concentrations with 0.2% L-arabinose into the medium.
Result

The result is quite clear. First, L-arabinose efficiently induced the expression of GFP. 0.2% arabinose seemed enough. However, D-arabinose did not have any effect. Secondly, glucose and trehalose suppressed the promoter, even though 0.2% L-arabinose was added. At the concentration of 0.3%, the effect of trehalose was even worse than that of glucose. The effect of glucose is well-known. However, that of trehalose is not. We give a hypothesis here. One trehalose can be hydrolyzed into two glucose. So 0.3% trehalose equals to 0.6% glucose. Thus, at a low concentration, trehalose is more insufferable to the promoter than glucose.
Description
Second, we measured the strength of the promoter by CAHS 106094 BBa_K3457012. This year, we used CAHS 106094 to protect bacteria from freeze-drying and dry storage. We used the promoter to express CAHS 106094 in E. coli DH5α strain to study whether CAHS 106094 worked in this strain.Protocol
The gene circuit we used is as below:

The protocol is as below:

【Day 1】Induction culture
(1) Pick clones which are in good condition and put them into 500 μL LB medium containing antibiotics. Shake them to
grow at 37℃ for 5~7 hours until the bacteria solution becomes turbid.
(2) Add 0.2% L-arabinose into 3 mL LB medium containing antibiotics. Add 3 μL of the bacteria solution mentioned in step 1
to dilute the bacteria by the ratio of 1:1000. Shake the solution to grow the bacteria at 37℃ overnight.
【Day 2】Freeze-dried
(1) If fluorescence induced by the iPTG is detectable in the control group (GFP), continue conducting the
experiment.
(2) Use spectrophotometer to measure the OD600 of the bacteria solution, OD600 = 1 equals to
109 cells. If the OD600 value is between 0.1 and 1, There is a linear relationship between
OD600 and bacterial density. Calculate the volume of bacterial solution for 109 cells by using
the formula V = 100 / (OD600 × Dilution ratio).
(3) Take out a measured amount of 109 cells and centrifuge it at 8000 rpm for 3 min. Then pour out the
supernatant.
(4) Resuspend the bacteria in a 15 mL tube with pre-refrigerated 100 μL 3% glucose solution.
(5) Take off the cover of the tube and put the bacteria into the cold trap. Open the compressor of the
lyophilization machine and freeze the shake tube for 2 h at -70℃.
(6) Put the caky bacteria solution into the drying chamber of the lyophilization machine. Open the vacuum pump to
dry it in vacuum for 6h at 1 Pa vacuum degree.
(7) Turn off the vacuum pump, place it at seal box filled with silica-gel desiccant a for 2 days at room
temperature.
【Day 3】Room temperature storage
【Day 4】Detect the survival rate
(1) Add 1 mL of sterile water to the tube, vortex for 15 s, placed it at room temperature for 10 min.
(2) Adjust the density of the bacteria solution by gradient dilution, then spread 100 μL of the bacteria solution on
the LB plate.
(3) If the density above is not suitable, take 100μL of the solution and spread it on the LB plate after several
gradient dilutions.
(4) Culture the bacteria overnight at 37℃.
【Day 5】Cell Count
(1) Take out the LB plate and take photos to record experimental results.
(2) Use the automatic cell counting function of Image J to count the colone number on the LB plate, then compare the
results between each group.
Result

As expected, the bacteria with an Arabinose promoter (BBa_I0500) and CAHS 106094 exhibited a higher survival rate, which indicated that the promoter and L-arabinose could successfully induce the expression of CAHS 106094 at enough level in E. coli DH5α strain..
Characterization by Jilin_China 2020
Validation of Arabinose response system
- See details in BBa_K3447103.
This part contains an arabinose operon, which constitutes the arabinose sensing system. By connecting the sfGFP gene downstream, we were able to verify the expression intensity of this arabinose operon.
To prove the arabinose sensing system can work successfully, a sfGFP was inserted after PBAD under the induction of arabinose at a series of concentrations. As shown in Fig. 1B, the expression efficiency was highest when induced with 0.2 g/mL arabinose.

Arabinose induces endotoxin expression
- See details in BBa_K3447104.
This part contains an arabinose operon, which constitutes the arabinose sensing system. By connecting the relE gene downstream, the normal growth and reproduction of bacteria will be inhibited when arabinose is present.
After the expression of arabinose operon was verified, sfGFP gene was replaced by relE gene, which causes the normal growth and reproduction of bacteria will be inhibited when there is arabinose in the medium.
The digestion and agarose gel electrophoresis are shown in Fig. 2.

Since relE encodes a small endotoxin peptide which represses the growth of bacteria, its expression could be characterized by measuring growth curve. Fig. 3A and 3B suggest that, compared with negative control, the E. coli cells cultured with standard arabinose can be inhibited for growth and reproduction. Finally, in order to verify whether the xylanase produced by E. coli could manage to hydrolyze xylan to arabinose and, by binding to the araC, turn on the expression of relE in the downstream of PBAD, thus proving its function by measuring the growth situation (Fig. 3C).

//direction/forward
//chassis/prokaryote/ecoli
//promoter
//regulation/positive
//classic/regulatory/other
| biology | |
| control | araC, arabinose |
| direction | Forward |
| n/a | Inducible pBad/araC promoter |
| negative_regulators | |
| o_h | |
| o_l | |
| positive_regulators | 1 |