
Difference between revisions of "Part:BBa K3781019"
| Line 75: | Line 75: | ||
<div><ul> | <div><ul> | ||
| − | <li style="display: inline-block;"> [[File:T--TU Kaiserslautern--Purification Ammoniumsulfat.png|thumb|none|600px|<b>Figure 5</b> | <b>Immunoblot</b> <html><a href="https://parts.igem.org/Part:BBa_K3781214">L1_sAP_RBD_GST</a></html> | after GST purification<br> | + | <li style="display: inline-block;"> [[File:T--TU Kaiserslautern--Purification Ammoniumsulfat.png|thumb|none|600px|<b>Figure 5</b> | <b>Immunoblot</b> | <html><a href="https://parts.igem.org/Part:BBa_K3781214">L1_sAP_RBD_GST</a></html> | after GST purification<br> |
1. AB | gt <b>anti-GST</b> | 1:10,000<br> | 1. AB | gt <b>anti-GST</b> | 1:10,000<br> | ||
2. AB | rb <b>anti-goat HRP</b> | 1:2,000<br> | 2. AB | rb <b>anti-goat HRP</b> | 1:2,000<br> | ||
Latest revision as of 21:09, 21 October 2021
GST + TEV Motif, MocloMania B5

This basic part codes for a GST-tag that is fused to an upstream TEV protease recognition motif.
The GST-tag has been used for purification purposes eversince the late 1980s and, unlike most other purification tags, doesn’t only consist of a small peptide, but rather an entire functional protein.[1] GST is short for glutathione S-transferase, a highly abundant cytosolic enzyme that is usually involved in protecting prokaryotic as well as eukaryotic cells from extracellular toxins.[2] It does this with the help of the redox peptide glutathione to whose reduced form it binds with very high affinity.[3] By fusing a gene of interest with a GST-tag, the resulting fusion protein can be purified from cell preparations with the help of affinity chromatography, employing reduced glutathione (GSH) as the respective column material. Since the affinity binding relies on the structural integrity of the GST protein, GST-tag purification can only be performed under native, non-denaturing conditions.[4]
The GST-tag encoded in this part is fused to a recognition site for the TEV protease, a cysteine protease domain originally derived from the tobacco etch virus, a plant virus that infects a wide variety of nightshades and weeds, including the eponymous tobacco plant.[5] This protease is known for its extremely high sequence specificity, meaning that it only cleaves proteins upon recognition of a fixed amino acid sequence, with little to no off-target effects.[6] When its motif is fused with a tag, it can be used for recovery of tag-free target protein after purification.
As a B5 part, this part is meant to occupy the most downstream position in the MocloMania cloning frame and is thus equipped with an additional stop codon.
size 26.4 kDa
function affinity purification tag
TEV recognition motif ENLYFQ/S
cloning position B5
plasmid backbone pAGM1301
Data
We were able to successfully clone this basic part into its respective L0 plasmid backbone and to confirm the integrity of the L0 construct via restriction digest and gel electrophoresis, see Figure 1. Furthermore, we were able to include it into a L1 construct, proving its correct adaptation towards MoClo assembly, see Figure 2.
-
 Figure 1 | Test digest of L0 B5 parts using BsaI
Figure 1 | Test digest of L0 B5 parts using BsaI
1 | pAGM1301 | 2247 + 598 bp
2 | L0_mCerulean_B5 | 2247 + 721 bp
3 | L0_mVenus_B5 | 2247 + 721 bp
4 | L0_GST_B5 | 2247 + 688 bp
L | Thermofischer GeneRuler Plus Ladder [bp] -
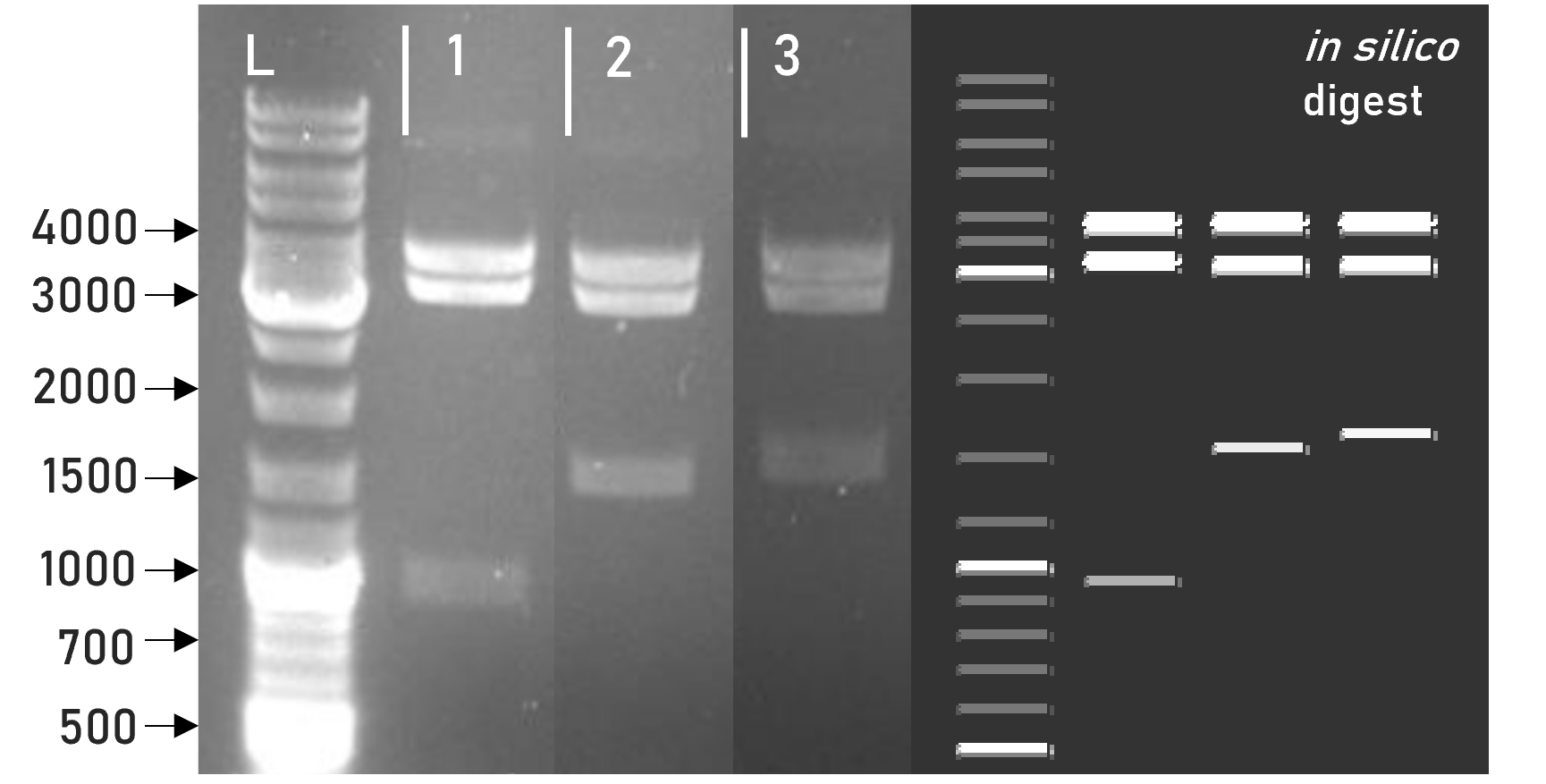 Figure 2 | Test digest of L1 constructs using HindIII
Figure 2 | Test digest of L1 constructs using HindIII
1 | L1_sAP_RBD_3xHA | 3792 + 3198 + 981 bp
2 | L1_sAP_RBD_GST | 3792 + 3198 + 1563 bp
3 | L1_sAP_mCerulean_GST | 3792 + 3198 + 1656 bp
L | Thermofischer GeneRuler Plus Ladder [bp]


Several different L1 constructs assembled with L0_GST_B5 have been successfully transfected into Leishmania and resulting recombinant protein expression could be observed via immunostaining on western blot, see Figures 3 and 4.
-
 Figure 3 | Immunoblot of L1 transfected Leishmania cell cultures | stained against RBD
Figure 3 | Immunoblot of L1 transfected Leishmania cell cultures | stained against RBD
1 | L1_sAP_RBD_Strep8His | 27.7 kDa
2 | L1_sAP_Myc_RBD_Strep8His | 30.2 kDa
3 | L1_sAP_RBD_GST | 51.9 kDa
n.c. | negative control | untransfected Leishmania culture
L | Thermofischer PageRuler Protein Ladder [kDa]
1. AB | ms anti-RBD | 1:2,000
2. AB | rb anti-ms HRP | 1:10,000 -
 Figure 4 | Immunoblot of L1 transfected Leishmania cell cultures | stained against GST
Figure 4 | Immunoblot of L1 transfected Leishmania cell cultures | stained against GST
1 | L1_sAP_RBD_GST | 51.9 kDa
2 | L1_sAP_Strep_mCerulean_GST | 55.9 kDa
n.c. | negative control | Leishmania culture transfected with empty L1 expression vector
L | Thermofischer PageRuler Protein Ladder [kDa]
1. AB | gt anti-GST | 1:10,000
2. AB | rb anti-gt HRP | 1:2,000


Both immunostaining against RBD, see Figure 3, as well as against the GST-tag, see Figure 4, showed recombinant protein expression for cultures transfected with L1_sAP_RBD_GST. This verifies the cohesive nature of our expressed fusion protein and proves the structural integrity of our GST-tag. In Figure 3, both the cell lysate as well as the culture supernatant of construct 3 | L1_sAP_RBD_GST display recombinant protein levels. In silico calculation predicts the full-length sAP_RBD_GST fusion protein to weigh about 55 kDa to which both upper bands can be attributed to. However, an additional, smaller band, running at approx. 27 kDa can be observed exclusively in the cell culture supernatant. Since this smaller band is also detected with anti-RBD antibodies, it is hypothesized that extracellular cleavage processes separate the RBD from its fusion tags after secretion of the fusion protein into the culture medium. For more information on this, please consult the sAP secretion tag part site.
In Figure 4, the situation is a little more convoluted. All the bands observed on this blot show the same general distribution pattern as other GST constructs, e.g. L1_sAP_RBD_mCerulean_GST, but the bands don't correspond with the predicted sizes of around 50 kDa and 27 kDa. We assume that this size shift is caused by technical deficiencies of the SDS-PAGE conduct. Shift of bands to unexplicably higher sizes might stem from a problem in gel density, caused for example by excess acrylamide concentrations. In construct 2 | L1_sAP_Strep_mCerulean_GST, the pattern of a higher band attributed to the full-length fusion protein and a smaller band around 27 kDa in the cell culture supernatant can be observed again. This time, tho, because we are staining against GST, the smaller band is assumed to correspond to cleaved molecular GST protein which is about 26 kDa in size. This explains why the smaller band can also be observed in the cell lysate, P and P+, especially for construct 1 | L1_sAP_RBD_GST, independently from secretion into the culture medium.
It also tells us that cleavage of the GST fusion tag is not only occurring extracellularly, but also intracellularly. The smeary bands in the supernatant of both constructs give rise to the idea that gradual degradation of the fusion protein might be occurring, maybe caused by uncoordinated proteolytic activity in the culture medium. This issue could be adversed by introducing protease inhibitor into the cell culture before protein harvest or analysis. We are working hard on optimizing expression levels and stability of fusion protein in order to make it even better accessible to downstream processing and increase yield.
The problem of fusion tag cleavage carries over into protein purification. Still, the C-terminal GST-tag remains our most successful purification tag. Several different L1 constructs assembled with L0_GST_B5 were shown to bind to gluthatione agarose resin during standard gravity-flow column purification, see Figure 5.
-
 Figure 5 | Immunoblot | L1_sAP_RBD_GST | after GST purification
Figure 5 | Immunoblot | L1_sAP_RBD_GST | after GST purification
1. AB | gt anti-GST | 1:10,000
2. AB | rb anti-goat HRP | 1:2,000
I Input | FT Flowthrough | W Wash | E Eluate | E.T Eluate, TCA precipitated
L.C | cell lysate of untransfected negative control
S.C | supernatant of untransfected negative control
S.A | supernatant after pelleting of ammonium precipitation
P.A | resuspended pellet of ammonium sulfate precipitation
GST | GST positive control | 26 kDa
RBD | RBD-GFP positive control | 52 kDa
L | Thermo Scientific PageRuler Prestained Protein Ladder [kDa] -
 Figure 6 | Immunoblot | L1_sAP_RBD_mVenus_GST | after GST-purification
Figure 6 | Immunoblot | L1_sAP_RBD_mVenus_GST | after GST-purification
1. AB | gt anti-GST | 1:10,000
2. AB | rb anti-goat HRP | 1:2,000
I Input | FT Flowthrough | W Wash | E Eluate | E.T Eluate, TCA precipitated
L | Thermo Scientific PageRuler Protein Ladder [kDa]


As can be seen in Figure 5, all samples of L1_sAP_RBD_GST, before and after application to the gravity-flow column, display two distinct protein bands with very high intensity. The upper band, running just under 55 kDa, can be attributed to the full-length fusion protein with an in silico size of 51.9 kDa. Due to its visualisation with anti-GST antibody and its running size of about 26 kDa, the lower band is suspected to consist of singular GST protein. This band pattern showing up across all lanes suggests a possible cleavage of the GST-tag from its fusion protein in the culture supernatant. This of course negatively affects purification yield, since recombinant RBD cannot be captured. Since the GST attributed band is also visible in flowthrough, FT, and wash, W, only partial binding of GST to the gluthatione agarose resin seems to occur. The affinity and specifity of binding is influenced by a wide variety of factors. In order to improve column binding, we are currently working hard on testing different paramters for pH conditions, temperature, buffer concentrations and procedural set-up. Even though singular GST tag is detected in all lanes with virtually identical intensity, the full-length protein seems to display increasing concentrations in the different elution steps, E and E.T, suggesting at least a partially successful purification of target protein.
In order to make secreted target proteins more accessible for purification procedures, it is often necessary to concentrate the cell culture supernatant, since secretion protein concentrations are low compared to cytosolic protein in cell lysates and since many purification systems have a limited applicable volume size. While our usual concentration process was done via centrifugation of the cell culture supernatant in AMICONs (CO 10,000 kDa), the purification in Figure 5 was conducted with a different concentration method, ammonium sulfate precipitation. This procedure was recommended to us by the scientific staff at JenaBioscience, the company distributing the cell culture kit that our expression vector is based on. Since protein purification for them gave good yields after ammonium sulfate precipitation, we wanted to implement this expert advice. As can be seen in S.A, the supernatant after spinning down the ammonium sulfate precipitate and P.A., the precipitate resuspended in buffer, substantial amounts of target protein could be carried over by this concentration procedure, whereas more singular GST seemed to remain partially solved.
In Figure 6, the GST-purification of culture transfected with L1_sAP_RBD_mVenus_GST shows three distinct protein bands in virtually all samples. Taking into consideration any reading or gel errors, the bands can be attributed to the full-length fusion protein (78,8 kDa), the GST-tag fused with the upstream mVenus (52 kDa) and the cleaved, singular GST tag (26 kDa). The band intensity in the flowthrough, FT, is almost similar to that in the supernatant input, I. This again suggests insufficient binding of the GST-tag to the gluthatione agarose column. Signal intensity is highest in the first washing step, W1, meaning that even if our fusion protein bound to the column, a large part of it is simply washed away and lost. Thus, we are working on optimizing the procedural set-up of purification by e.g. decreasing wash buffer volume and composition. This way, we hope to facilitate efficient purification of L1 constructs from Leishmania tarentolae cell culture, so more researchers can implement it for their protein production needs.
The MocloMania collection
This basic part is part of the MocloMania collection, the very first collection of genetic parts specifically designed and optimized for Modular Cloning assembly and recombinant protein expression in the protozoan parasite Leishmania tarentolae.
Are you trying to express complexly glycosylated proteins? Large antibody side chains? Human proteins that require accurate post-translational modification? Then Leishmania might be just the right organism for you! Leishmania tarentolae’s glycosylation patterns resemble those of human cells more closely than any other microbial expression host, while still delivering all the benefits of microbial production systems like easy transfection and cultivation.[7] So instead of relying on mammalian cell lines, try considering Leishmania as your new expression host of choice!
Our MocloMania collection will allow you to easily modify your protein of choice and make it suitable for downstream detection and purification procedures - all thanks to the help of Modular Cloning. This cloning system was first established by Weber et al. in 2011 and relies on the ability of type IIS restriction enzymes to cut DNA outside of their recognition sequence, hereby generating four nucleotide overhangs.[8] Every basic part in our collection is equipped with a specified set of overhangs that assign it to its designated position within the reading frame. These so-called cloning positions are labelled B2-B5 from upstream to downstream. By filling all positions with the basic parts of your choice, you can easily generate variable genetic constructs that code for the fusion protein of your desire.
We furthermore provide a specifically domesticated Leishmania expression vector, named weird_plex, which will package your fusion construct into a functional transcriptional unit that is optimized for high expression in Leishmania.
The best part? Because of the type IIS restriction properties and the specifity of the generated overhangs, restriction and ligation of your construct can all happen simultaneously in a simple one-step, one-pot reaction. This will safe you a lot of time and frustration in your cloning endeavours!
Do we have your attention? In the table below you can find some basic information on how our cloning system, along with most other MoClo systems, is set up. Please feel free to check out our wiki to find more information on Leishmania and Modular Cloning as well as to understand how this basic part integrates into our part collection. See you there!
| Level | What does this level contain? | antibiotic resistance | Enzyme used for ligation |
| L0 | The foundation to every MoClo construct which are basic genetic units, such as coding sequences, promoters, terminators | spectinomycin | BbsI |
| L1 | Several L0 parts assembled into a functional transcriptional unit, e.g. consisting of promoter, coding region and terminator | ampicillin | BsaI |
| L2 | Multiple transcriptional units added into one multi-gene construct, e.g. a protein of interest fused to a selection marker | kanamycin | BbsI |
Sequence and Features
IMPORTANT
This basic part has been altered in its sequence in order to make it compatible with the iGEM RCF 1000 cloning standard.
In its original form, this part contains two endogenous SapI restriction sites. This is because it was specifically designed to be utilized within the Modular Cloning system as established by Weber et al. in 2011, relying on BsaI and BbsI as the executive type IIS restriction enzymes.[9] This MoClo standard is a very frequently used type IIS standard, especially when it comes to plant-based expression systems.[10][11]
Because of its intended use within the MoClo system, this basic part is domesticated against BsaI and BbsI, but not against SapI. Upon registration of this part in the iGEM Registry, the endogenous SapI restriction sites were disrupted via in silico introduction of a silent mutation. We decided to do this in order to increase the chance of the judges actually taking a look at this part page and not having the part overlooked due to incompatibility.
Since the part’s amino acid sequence remains unchanged, users interested in utilizing this part for type IIS loop assembly can rely on the sequence as listed on this page. For users interested in applying this basic part to their MoClo based cloning approaches, in silico reversion of the mutations might be beneficial, since SapI proved to be a valuable enzyme for confirming part insertion via restriction digest.
We highly recommend that researchers and iGEM teams utilize this basic part within the MocloMania cloning system, since it is codon optimized towards Leishmania tarentolae and may otherwise negatively influence expression yield in other organisms.
MUTATION 1 | bp 604 - 606 | AGC → TCG
MUTATION 2 | bp 676 - 678 | AGC → TCG
- 10INCOMPATIBLE WITH RFC[10]Illegal PstI site found at 628
- 12INCOMPATIBLE WITH RFC[12]Illegal PstI site found at 628
- 21COMPATIBLE WITH RFC[21]
- 23INCOMPATIBLE WITH RFC[23]Illegal PstI site found at 628
- 25INCOMPATIBLE WITH RFC[25]Illegal PstI site found at 628
- 1000COMPATIBLE WITH RFC[1000]
Reference Literature
- ↑ Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988 Jul 15;67(1):31-40. doi: 10.1016/0378-1119(88)90005-4. PMID: 3047011.
- ↑ Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51-88. doi: 10.1146/annurev.pharmtox.45.120403.095857. PMID: 15822171.
- ↑ Lohning, Anna, Salinas, Anna, Wong, M.G., 1999/04/01, Glutathione S-transferases--a review, Current Medicinal Chemistry
- ↑ Harper S, Speicher DW. Purification of proteins fused to glutathione S-transferase. Methods Mol Biol. 2011;681:259-280. doi:10.1007/978-1-60761-913-0_14
- ↑ L. L. Breman (July 1987). "Tobacco Etch Virus" (PDF). Plant Pathology Circular No. 297 Fla. Dept. Agric. & Consumer Serv. Retrieved July 12, 2017.
- ↑ Kapust RB, Waugh DS (July 2000). "Controlled intracellular processing of fusion proteins by TEV protease". Protein Expr. Purif. 19 (2): 312–8. doi:10.1006/prep.2000.1251. PMID 10873547.
- ↑ Langer T, Corvey C, Kroll K, Boscheinen O, Wendrich T, Dittrich W. Expression and purification of the extracellular domains of human glycoprotein VI (GPVI) and the receptor for advanced glycation end products (RAGE) from Rattus norvegicus in Leishmania tarentolae. Prep Biochem Biotechnol. 2017 Nov 26;47(10):1008-1015. doi: 10.1080/10826068.2017.1365252. Epub 2017 Aug 31. PMID: 28857681.
- ↑ Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S (2011) A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 6(2): e16765. https://doi.org/10.1371/journal.pone.0016765
- ↑ Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S (2011) A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLoS ONE 6(2): e16765. https://doi.org/10.1371/journal.pone.0016765
- ↑ Patron, N.J., Orzaez, D., Marillonnet, S. et al., (2015), Standards for plant synthetic biology: a common syntax for exchange of DNA parts. New Phytol, 208: 13-19. https://doi.org/10.1111/nph.13532
- ↑ https://www.addgene.org/cloning/moclo/, last visited 10/16/21, 19:00 ECT