
Part:BBa_K3001003
Cas13a Lbu + T7 promoter
This protein is an RNA cleaving enzyme called Cas13a that was isolated from Leptotrichia buccalis (Lbu). It interacts with a direct repeat stem loop on a CRISPR RNA (crRNA) that contains a specific sequence for a target RNA transcript. This construct is designed to be able to be expressed under an inducible promoter (T7). The protein itself has several purification tags (His, Twin strep tag, and a SUMO tag). This construct was ordered from Addgene.
Wetlab Data
Both parts of our system rely on the protein Cas13a, so it was imperative that we purify the protein for future use in our Cas13a activity assay. Our team successfully purified Lbu Cas13a protein while we were unable to purify Lwa Cas13a to a high enough concentration through Nickel Affinity Chromatography and Size Exclusion Chromatography. Initially, our team tried to express Cas13a in E. coli BL21 DE3; however, we had issues with overexpression so we tried again using E. coli Rosetta DE3 cells and saw greater success (Figure 1).
 Figure 1. 10% SDS PAGE of overexpression of Lbu and Lwa Cas13a. Run at 180 V for 25 minutes and 200 V for 2.5 hours. Left to right: lane 1: Lbu before induction; lane 2: Lbu after induction; lane 3: Lbu before induction; lane 4: Lbu after induction; lane 5: Lwa before induction; lane 6: Lwa after induction; lane 7: Lwa before induction; lane 8: Lwa after induction; lane 9: 10-250 kDa ladder; lane 10: empty.
Figure 1. 10% SDS PAGE of overexpression of Lbu and Lwa Cas13a. Run at 180 V for 25 minutes and 200 V for 2.5 hours. Left to right: lane 1: Lbu before induction; lane 2: Lbu after induction; lane 3: Lbu before induction; lane 4: Lbu after induction; lane 5: Lwa before induction; lane 6: Lwa after induction; lane 7: Lwa before induction; lane 8: Lwa after induction; lane 9: 10-250 kDa ladder; lane 10: empty.
Those cells were lysed then centrifuged to separate the supernatant and cell pellet. The lysate was then introduced to a Nickel Sepharose affinity column to isolate the Cas13a protein. It was bound to the Nickel Sepharose due to the histidine tag.
 Figure 2. 10% SDS PAGE of Lbu Cas13a protein after his tag purification. Left to right: lane 1: Lbu lysate; lane 2: Lbu lysate post-binding; lane 3: Wash (pooled); lane 4: elution 1; lane 5: elution 2; lane 6: elution 3; lane 7: elution 4; lane 8: elution 5; lane 9: elution 6; lane 10: 10-250 kDa ladder.
Figure 2. 10% SDS PAGE of Lbu Cas13a protein after his tag purification. Left to right: lane 1: Lbu lysate; lane 2: Lbu lysate post-binding; lane 3: Wash (pooled); lane 4: elution 1; lane 5: elution 2; lane 6: elution 3; lane 7: elution 4; lane 8: elution 5; lane 9: elution 6; lane 10: 10-250 kDa ladder.
To further purify Cas13a, we ran the partially purified protein solution through a column, that contained beads. The beads have small crevices and this causes proteins of different sizes to pass through during different times. Again, the band for Lwa did not show up well after scanning the gel. Since it was likely that a low amount of protein was present in our collected fractions, we chose to move forward only with our purified Lbu. Multiple bands appeared in our purified Lbu Cas13a protein, and further purification steps would aid in ensuring the accuracy of in vitro data in the future. However, compared to previous purification data from [http://2017.igem.org/Team:Munich/Cas13a Munich 2017], we are working with a much purer sample than has been used in the competition.
 Figure 3. Purification of Lbu Cas13a using size exclusion chromatography. (A) 10% SDS PAGE of Lbu Cas13a after size exclusion chromatography. Left to right: lane 1: Lbu concentrated after affinity chromatography; lane 2: Lbu frac H2; lane 3: Lbu frac H1; lane 4: Lbu frac I1; lane 5: Lbu frac I2; lane 6: Lbu frac I3; lane 7: Lbu frac I4; lane 8: Lbu frac I6; lane 9: Lbu post-SEC and concentrated; lane 10: 10-250 kDa ladder. (B) Chromatogram generated by AktaPrime of Lbu Cas13a purified using a large Superdex75 column (GE Life Sciences).
Figure 3. Purification of Lbu Cas13a using size exclusion chromatography. (A) 10% SDS PAGE of Lbu Cas13a after size exclusion chromatography. Left to right: lane 1: Lbu concentrated after affinity chromatography; lane 2: Lbu frac H2; lane 3: Lbu frac H1; lane 4: Lbu frac I1; lane 5: Lbu frac I2; lane 6: Lbu frac I3; lane 7: Lbu frac I4; lane 8: Lbu frac I6; lane 9: Lbu post-SEC and concentrated; lane 10: 10-250 kDa ladder. (B) Chromatogram generated by AktaPrime of Lbu Cas13a purified using a large Superdex75 column (GE Life Sciences).
We noticed a large peak at absorbance 254 nm, which is the level at which nucleic acids absorbs. Since Cas13a binds and interacts with RNA this is likely what is causing the peak. We ran the sample on a urea page to confirm the presence of RNA (Figure 4). Members of the Wieden lab at the University of Lethbridge have worked with proteins that interact with RNA. To minimize these interactions, they purify this protein using anion exchange chromatography (Q Sepharose). We noticed that previous iGEM teams had not done this purification step in the past. To minimize any issues with other interacting RNAs, we recommend future iGEM teams complete this purification step in between nickel affinity chromatography and size exclusion chromatography. Due to time constraints, we were unable to test this purification methodology to see if it impacted our results.
 Figure 4. 10% Urea PAGE of Lbu and Lwa protein samples after affinity and size exclusion chromatography purification. Left to right: lane 1: High Range RiboRuler; lane 2: Lwa sample post-concentration after affinity purification; lane 3: Lbu sample post-concentration after size exclusion chromatography; lane 4: Lbu sample post-concentration after affinity chromatography.
Figure 4. 10% Urea PAGE of Lbu and Lwa protein samples after affinity and size exclusion chromatography purification. Left to right: lane 1: High Range RiboRuler; lane 2: Lwa sample post-concentration after affinity purification; lane 3: Lbu sample post-concentration after size exclusion chromatography; lane 4: Lbu sample post-concentration after affinity chromatography.
Our team conducted a Cas13a activity assay to test the effectiveness of our enzyme. We incubated 300 nM of Lbu Cas13a complexed with the crRNA with various concentrations of RNA Mango to see the change in fluorescence of the dye thiazole orange. By analyzing our results, we can see that the fluorescence for the RNA Mango at the concentration 25nM sample decreased. This likely indicates that the Cas13a enzyme is cleaving. However, for higher concentrations, there was not a significant change in fluorescence. This may be due to having an insufficient amount of enzyme for proper cleaving to occur or our enzyme not being active enough to cleave larger amounts of RNA Mango in the same amount of time as the 25 nM sample. The sample with concentration 100 nM is excluded from these generalizations. We believe there may have been an error made in the reading. Another reason for the differences seen in the concentrations of RNA Mango could be caused by if our purified RNA is taking on multiple conformations that affect its ability to interact with thiazole orange. If the G Quadruplex is not forming properly, we would not see fluorescence. Graph C shows the results of our control experiment. Similar to the previously mentioned experiment we incubated 300 nM of Lbu Cas13a complexed with the crRNA with various concentrations of RNA, however this time we used RybA. This was a negative control experiment to test the specificity of our CRISPR Cas13a system. It seems that there was no significant change in fluorescence over time thereby indicating that no RybA was cleaved and our CRISPR Cas13a is specific. Since there was no added fluorescence molecule, it is also hard to interpret what any changes would be.
 Figure 5. Determining activity of Lbu Cas13a by targeting snR30 containing RNA Mango II by detecting a loss of fluorescence. Excitation occurred at 510 nm and emission at 535 nm and scans were completed for 3 hours. Data was normalized by dividing by the negative control, which contained all components except for RNA. (A) Raw scans normalized to the negative control of snR30-RNA Mango II at various concentrations in complex with Lbu Cas13a and the crRNA (n=1). Controls are also shown of only snR30-RNA Mango II, the target molecule and Lbu Cas13a, and the target molecule and RNase A (n=2 +/- SD for controls). (B) Relative fluorescence at the maximum fluorescence of snR30-RNA Mango II at 72 minutes (n=1 for snR30-RNA Mango II at various concentrations, n=2 +/- SD for controls). (C) Controls for activity of Lbu Cas13a and crRNA. RybA was used as a specificity control for cleavage (n=2 +/- SD for all data shown).
Figure 5. Determining activity of Lbu Cas13a by targeting snR30 containing RNA Mango II by detecting a loss of fluorescence. Excitation occurred at 510 nm and emission at 535 nm and scans were completed for 3 hours. Data was normalized by dividing by the negative control, which contained all components except for RNA. (A) Raw scans normalized to the negative control of snR30-RNA Mango II at various concentrations in complex with Lbu Cas13a and the crRNA (n=1). Controls are also shown of only snR30-RNA Mango II, the target molecule and Lbu Cas13a, and the target molecule and RNase A (n=2 +/- SD for controls). (B) Relative fluorescence at the maximum fluorescence of snR30-RNA Mango II at 72 minutes (n=1 for snR30-RNA Mango II at various concentrations, n=2 +/- SD for controls). (C) Controls for activity of Lbu Cas13a and crRNA. RybA was used as a specificity control for cleavage (n=2 +/- SD for all data shown).
To confirm the effectiveness of our Cas13a enzyme we ran before and after fluorescence scanning samples on a urea page. Additionally, this would allow us to better interpret our experimental controls. This confirmed our speculation that the 25 nM sample was indeed cleaved by Lbu Cas13a. For the other samples, no significant difference is seen between the before and after samples. For the 50 nM before sample, we believe there may have been a loading issue that affected the quantification results seen in table 1. Our controls seen in Figure 6 B and C show that there are no significant changes between before and after scanning the samples. This means that are assay is specific for our targeted molecule needing to be present to have enzyme activation. Cas13a will also not cleave our target molecule without the crRNA in the complex.
 Figure 6. 10% Urea PAGEs of Lbu Cas13a activity assay components. (A) Left to right: lanes 1-3: empty; lane 4: RNA Mango 100 nM post-scan; lane 5: RNA Mango 100 nM pre-scan; lane 6: RNA Mango 75 nM post-scan; lane 7: RNA Mango 75 nM pre-scan; lane 8: RNA Mango 50 nM post-scan; lane 9: RNA Mango 50 nM pre-scan; lane 10: RNA Mango 25 nM post-scan; lane 11: RNA Mango 25 nM pre-scan; lane 12: RNA Mango + RNase A post-scan; lane 13: RNA Mango + RNase A pre-scan; lane 14: negative control (no target or control RNA) post-scan; lane 15: negative control (no target or control RNA) pre-scan. (B) Left to right: lane 1: RNase A + RybA pre-scan; lane 2: RNase A + RybA post-scan; lane 3: RybA 25 nM pre-scan; lane 4: RybA 25 nM post-scan; lane 5: RybA 50 nM pre-scan; lane 6: RybA 50 nM post-scan; lane 7: RybA 75 nM pre-scan; lane 8: RybA 75 nM post-scan; lane 9: RybA 100 nM pre-scan; lane 10: RybA 100 nM post-scan.
(C) Left to right: lane 1: crRNA pre-scan; lane 2: crRNA post-scan; lane 3: RNA Mango pre-scan; lane 4: RNA Mango post-scan; lane 5: RybA pre-scan; lane 6: RybA post-scan; lane 7: Cas13a Lbu pre-scan; lane 8: Cas13a Lbu post-scan; lane 9: Cas13a Lbu + crRNA pre-scan; lane 10: Cas13a Lbu + crRNA post-scan; lane 11: Cas13a Lbu + RNA Mango pre-scan; lane 12: Cas13a Lbu + RNA Mango post-scan; lane 13: Cas13a Lbu + RybA pre-scan; lane 14: Cas13a Lbu + RybA post-scan. Lane 15: empty.
Figure 6. 10% Urea PAGEs of Lbu Cas13a activity assay components. (A) Left to right: lanes 1-3: empty; lane 4: RNA Mango 100 nM post-scan; lane 5: RNA Mango 100 nM pre-scan; lane 6: RNA Mango 75 nM post-scan; lane 7: RNA Mango 75 nM pre-scan; lane 8: RNA Mango 50 nM post-scan; lane 9: RNA Mango 50 nM pre-scan; lane 10: RNA Mango 25 nM post-scan; lane 11: RNA Mango 25 nM pre-scan; lane 12: RNA Mango + RNase A post-scan; lane 13: RNA Mango + RNase A pre-scan; lane 14: negative control (no target or control RNA) post-scan; lane 15: negative control (no target or control RNA) pre-scan. (B) Left to right: lane 1: RNase A + RybA pre-scan; lane 2: RNase A + RybA post-scan; lane 3: RybA 25 nM pre-scan; lane 4: RybA 25 nM post-scan; lane 5: RybA 50 nM pre-scan; lane 6: RybA 50 nM post-scan; lane 7: RybA 75 nM pre-scan; lane 8: RybA 75 nM post-scan; lane 9: RybA 100 nM pre-scan; lane 10: RybA 100 nM post-scan.
(C) Left to right: lane 1: crRNA pre-scan; lane 2: crRNA post-scan; lane 3: RNA Mango pre-scan; lane 4: RNA Mango post-scan; lane 5: RybA pre-scan; lane 6: RybA post-scan; lane 7: Cas13a Lbu pre-scan; lane 8: Cas13a Lbu post-scan; lane 9: Cas13a Lbu + crRNA pre-scan; lane 10: Cas13a Lbu + crRNA post-scan; lane 11: Cas13a Lbu + RNA Mango pre-scan; lane 12: Cas13a Lbu + RNA Mango post-scan; lane 13: Cas13a Lbu + RybA pre-scan; lane 14: Cas13a Lbu + RybA post-scan. Lane 15: empty.
Table 1. Quantification Figure 6 A lanes 4-11 of decrease in RNA Mango using ImageJ.
| Concentration of RNA Mango (nM) | 25 | 50 | 75 | 100 |
|---|---|---|---|---|
| Pre-scan | 49.34% | 20.8% | 45.3% | 50.0% |
| Post-scan | 1.32% | 79.2% | 54.5% | 50.0% |
Initially, our team had planned on doing a triple plasmid transformation of our target fluorescent protein (GFP), crRNA, and Cas13a to test if our system would work in vivo . Additionally, we wanted to have RFP as the fluorescent protein to serve as a specificity control instead of transforming the plasmid containing GFP. We were unsuccessful in getting all three plasmids to transform, but did succeed in getting the fluorescent proteins and crRNA containing plasmids to transform together.
As an alternative experiment, our team grew cells that expressed the Lbu and Lwa Cas13a protein overnight. We also grew cells that expressed dual plasmids; GFP and crRNA Lwa; GFP and crRNA Lbu; RFP and crRNA Lwa; and RFP and crRNA Lbu. We then lysed the cells that expressed the Cas13a proteins using a French Press and clarified the lysate via centrifugation. Following this, our team pipetted in a 1:1 ratio of clarified cell lysate: fluorescent protein and crRNA into a 96 well plate. This allowed us to observe if there would be an effect from the CRISPR Cas13a system on the fluorescent proteins. We observed that in our optical density data, both dual plasmid systems for GFP and RFP had stunted growth in comparison to only E. coli cells expressing GFP or RFP or no plasmid (Figure 7C). Adding the lysate may have caused the death of the culture. We neglected to include replicates of the dual plasmid system without adding lysate to observe how that grew. This would be beneficial for any future experiments. Alternatively, there may have been some effect of the protein in the lysate on the GFP fluorescence (Figure 7B). However, we are unsure of the specificity due to the potential of the RFP not folding correctly in vivo as demonstrated by the substantial standard deviation seen in our replicates (Figure 7A).
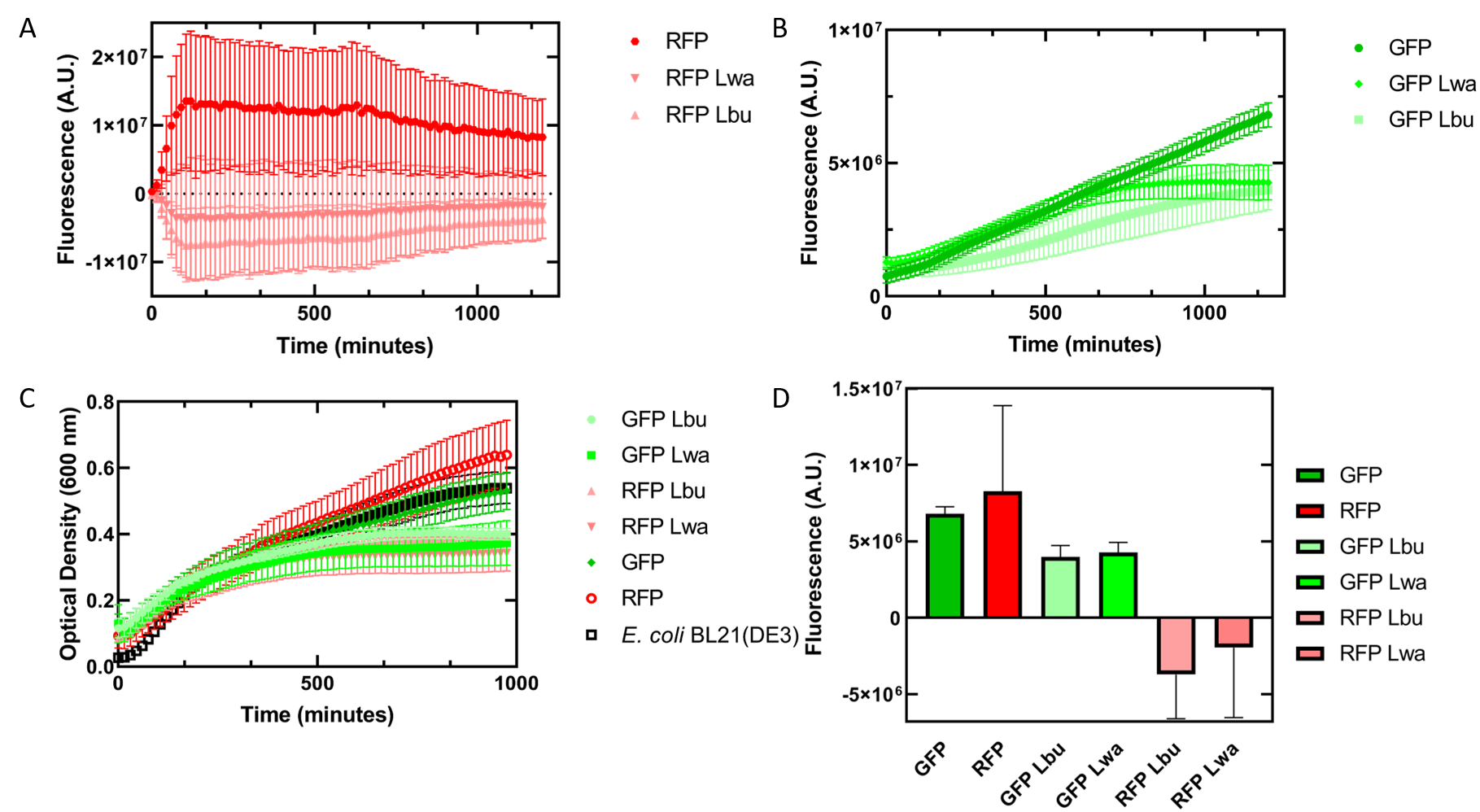 Figure 7. In vivo fluorescence assay of E. coli BL21(DE3) cells containing fluorescent protein and crRNA plasmids and E. coli Rosetta(DE3) cell lysate of overexpressed Cas13a proteins. This assay was conducted with 3 biological replicates and 3 technical replicates. (A) Fluorescence of E. coli cells containing only an RFP expressing plasmid, or dual plasmid expression of RFP and crRNAs from Lwa and Lbu that target GFP and the respective cell lysate containing the appropriate Cas13a. RFP excitation was at 558 nm and emission at 583 nm. (B) Fluorescence of E. coli cells containing only a GFP expressing plasmid, or dual plasmid expression of GFP and crRNAs from Lwa and Lbu that target GFP and the respective cell lysate containing the appropriate Cas13a. GFP excitation was at 475 nm and emission at 508 nm. (C) Optical density of E. coli cells expressing GFP, RFP, dual plasmid systems mentioned previously, or only E. coli BL21(DE3) cells with absorbance measured at 600 nm. (D) Relative fluorescence at maximum excitation at 81 minutes. GFP excitation was at 475 nm and emission at 508 nm and RFP excitation was at 558 nm and emission at 583 nm.
Figure 7. In vivo fluorescence assay of E. coli BL21(DE3) cells containing fluorescent protein and crRNA plasmids and E. coli Rosetta(DE3) cell lysate of overexpressed Cas13a proteins. This assay was conducted with 3 biological replicates and 3 technical replicates. (A) Fluorescence of E. coli cells containing only an RFP expressing plasmid, or dual plasmid expression of RFP and crRNAs from Lwa and Lbu that target GFP and the respective cell lysate containing the appropriate Cas13a. RFP excitation was at 558 nm and emission at 583 nm. (B) Fluorescence of E. coli cells containing only a GFP expressing plasmid, or dual plasmid expression of GFP and crRNAs from Lwa and Lbu that target GFP and the respective cell lysate containing the appropriate Cas13a. GFP excitation was at 475 nm and emission at 508 nm. (C) Optical density of E. coli cells expressing GFP, RFP, dual plasmid systems mentioned previously, or only E. coli BL21(DE3) cells with absorbance measured at 600 nm. (D) Relative fluorescence at maximum excitation at 81 minutes. GFP excitation was at 475 nm and emission at 508 nm and RFP excitation was at 558 nm and emission at 583 nm.
The 2021 Lethbridge iGEM Team designed an improved version of this part, which includes an N-terminal 6xHistidine tag for purification and a C-terminal anionic tag for encapsulation by the MS2 phage-like particle. This improved part can be found here: BBa_K3738023
Sequence and Features
- 10INCOMPATIBLE WITH RFC[10]Illegal PstI site found at 1564
Illegal PstI site found at 2419
Illegal PstI site found at 3358 - 12INCOMPATIBLE WITH RFC[12]Illegal NheI site found at 107
Illegal PstI site found at 1564
Illegal PstI site found at 2419
Illegal PstI site found at 3358 - 21INCOMPATIBLE WITH RFC[21]Illegal BglII site found at 297
Illegal BglII site found at 845
Illegal BglII site found at 1781
Illegal BglII site found at 1991
Illegal BglII site found at 2045
Illegal BglII site found at 2276
Illegal BglII site found at 2549 - 23INCOMPATIBLE WITH RFC[23]Illegal PstI site found at 1564
Illegal PstI site found at 2419
Illegal PstI site found at 3358 - 25INCOMPATIBLE WITH RFC[25]Illegal PstI site found at 1564
Illegal PstI site found at 2419
Illegal PstI site found at 3358
Illegal NgoMIV site found at 2497
Illegal NgoMIV site found at 3133 - 1000COMPATIBLE WITH RFC[1000]
| None |