
Difference between revisions of "Part:BBa K143013"
m |
|||
| (48 intermediate revisions by 4 users not shown) | |||
| Line 1: | Line 1: | ||
__NOTOC__ | __NOTOC__ | ||
<partinfo>BBa_K143013 short</partinfo> | <partinfo>BBa_K143013 short</partinfo> | ||
| + | ===<big>Promoter 43: Constitutive Promoter for ''B. subtilis''</big>=== | ||
Promoter 43 is a constitutive promoter that constitutively expresses the P43 protein in ''B.subtilis''. This promoter has been shown to be recognized and active during the exponential and lag phases of growth. It has been hypothesized that the ability to recognize the promoter in exponential and lag phase of growth is due to the recognition of the promoter by both sigma factor 55 (the major sigma factor A) and sigma factor 37 (the lag phase sigma factor B) <cite>1</cite>. The P43 promoter has been previously used for constitutive expression of exogenous genes within ''B.subtilis'' vectors <cite>2</cite>. The context with which we used the promoter P43 is as a '''Polymerase Per Second''' (PoPS) generator. | Promoter 43 is a constitutive promoter that constitutively expresses the P43 protein in ''B.subtilis''. This promoter has been shown to be recognized and active during the exponential and lag phases of growth. It has been hypothesized that the ability to recognize the promoter in exponential and lag phase of growth is due to the recognition of the promoter by both sigma factor 55 (the major sigma factor A) and sigma factor 37 (the lag phase sigma factor B) <cite>1</cite>. The P43 promoter has been previously used for constitutive expression of exogenous genes within ''B.subtilis'' vectors <cite>2</cite>. The context with which we used the promoter P43 is as a '''Polymerase Per Second''' (PoPS) generator. | ||
| + | |||
| + | |||
| + | |||
| + | ''' | ||
| + | ==KEYSTONE 2022 Characterization== | ||
| + | Promoter 43 was utilized with the gene coded for the over-expression of degQ, which has been knocked into the B. subtilis to produce the cyclolipopeptide fengycin. | ||
| + | ===Fengycins production in <i>Bacillus subtilis</i> 168=== | ||
| + | The main composite of our product, fengycins, is produced by engineering <i>Bacillus subtilis</i> 168. As it is shown in the demonstration in description, sfp and degQ genes are critical to the biosynthesis of fengycins. Besides, <i>Bacillus subtilis</i> 168 possesses a natural but invalid sfp gene, and the functional degQ gene is silenced with the regulation of a weak promoter.Thus, with the help of pJOE8999_sfp_degQ plasmid (Figure 1a), we knocked in both sfp gene and degQ gene in the region of the natural sfp gene of <i>Bacillus subtilis</i> 168 after being knocking out. Under the guidance of protocol, we successfully transformed and induced the plasmid to function as a gene editor (Figure 1c). After induction, 7 strains were selected for PCR and electrophoresis verification. Since successful knock-in would would result in a 252 bps increase in the genome of <i>Bacillus subtilis</i> 168, the electrophoresis result conveys preliminarily that all of these strains has achieved our target of constructing the fengycins producers (Figure 1b). | ||
| + | [[Image:Parts-keystone-fengycin1.jpeg|thumbnail|750px|center|'''Figure 1:''' | ||
| + | Edting the genome of <i>Bacillus subtilis</i> 168 to enable it to produce fengycins. (a) Plasmid design for knocking out the invalid sfp gene of <i>Bacillus subtilis</i> 168 and knocking in the sfp gene from B. amyloliquefaciens FZB42 and degQ gene from <i>Bacillus subtilis</i> 168. (b) Electrophoresis results show that gene editing is successful. (c) Protocol about transformation, induction and elimination of pJOE8999 plasmid in <i>Bacillus subtilis</i> 168. ]] | ||
| + | After sequencing further verified that the successful construction of the fengycins producing strain, we used the strain for fermentation (Figure 2a). For the quantification of fengycins production, the methanol - extract obtained from the fermentation broth was analyzed by high performance liquid chromatography (HPLC). The peak of fengycins appeared at the retention time of 8-11.5 min under methanol mobile phase (Figure 2c). The injection sample was a solution of lipopeptide extract from 30 mL of fermentation broth dissolved in 7 mL methanol. Besides, various concentrations of fengycins standard is also injected for the construction of the standard curve of peak area obtained by HPLC (y) and its corresponding fengycins’ concentration (x). Based on the standard curve, we got the regression curve and its corresponding formula y=9.767x+193.44, and R2>0.98 displays that the regression equation fits the observed values (Figure 2b). Substituting y=1242.3 obtained from the sample into the regression equation, the concentration of fengycin in methanol solution can be calculated as 107.4 mg/L, that is, 25.06 mg/L of fengycins can be achieved by shaking-flask fermentation. | ||
| + | [[Image:Parts-keystone-fengycin2-1.jpeg|750px|thumbnail|center|'''Figure 2:''' | ||
| + | Quantification analysis of fengycins production. (a) The sequencing result of engineering <i>Bacillus subtilis</i> 168 strain for production. (b) The standard curve of peak area obtained by HPLC (y) and its corresponding fengycins' concentration (x). (c) HPLC results of sample and various concentrations of fengycins standard. ]] | ||
| + | |||
| + | |||
| + | ===BSC_United's Characterization to Promoter 43 === | ||
| + | Promoter P43 is a strong promoter of ''Bacillus subtilis'' expression system. It is derived from cytidine deaminase (cdd) gene in ''B. subtilis ''. It is often used to express different target proteins. | ||
| + | Several promoters with high expression level that are commonly used in ''Bacillus subtilis'' were screened to determine the most efficient ones for our experiments on the construction of biological parts. Three of such promoters, Groe, SecA, P43, exist in the iGEM registry already. | ||
| + | The primer sequences for individual promoters used for our synthetic biology project are as follows: | ||
| + | |||
| + | groe-up CGCGGATCCCCAATACTGTTTTCTCAAATGGTATGTA | ||
| + | |||
| + | groe-down CGGGGTACCTGAAATAACCTCCTCAATAGTATGA | ||
| + | |||
| + | YxiE-up CGCGGATCCAATTGAAGCGCGCGAAGCCA | ||
| + | |||
| + | YxiE-down CGGGGTACCGCTCTTCCCGCCTTTCGGACTG | ||
| + | |||
| + | SecA-up CGCGGATCCGGACATCGTCCGTCAGAAACGCTTT | ||
| + | |||
| + | SecA-down CGGGGTACCTCACACGCCTATTTTAGAGGCATGT | ||
| + | |||
| + | Ylbp-up CGCGGATCCGTCACAACAGTCACGTCGTGATAAA | ||
| + | |||
| + | Ylbp-down CGGGGTACCACATATATTGTAAACGCTTTATTTA | ||
| + | |||
| + | P43-up CGCGGATCCTGATAGGTGGTATGTTTTCG | ||
| + | |||
| + | P43-down CGGGGTACCTATAATGGTACCGCTATCACT | ||
| + | |||
| + | === Strains and Plasmids === | ||
| + | <p>The bacterium strain we used was B. subtilis WB600, and the plasmid was WB0911H-ASN (with the antibiotic ampicillin and kanamycin resistance cassette). Both the strain and plasmid were kindly donated to us by Professor Jian CHEN at Jiannan University, China.</p> | ||
| + | |||
| + | <html> | ||
| + | <center> | ||
| + | <figure> | ||
| + | <img width="70%" src="https://2019.igem.org/wiki/images/8/83/T--BSC_United--PartI_plasmid_map.png"> | ||
| + | </figure> | ||
| + | <h5>Figure 1. pMAA0911H-ASN transformation vector</h5> | ||
| + | </center> | ||
| + | </html> | ||
| + | <br/> | ||
| + | |||
| + | <P>The promoters Groe (BBa J100034), SecA (BBa K1469002), and P43 (BBa K143013) were used to replace the original promoter hpall in WB0911H-ASN respectively. Here ASN stands for L-asparaginase (EC.3.5.1.1). By the comparison of the ASN activity expressed in the transformants, we found that the p43 promoter was the strongest promoter. Therefore, it was selected for our project.</p><br> | ||
| + | The promoters Groe (BBa J100034), SecA (BBa K1469002), and P43 (BBa K143013) were used to replace the original promoter hpall in WB0911H-ASN respectively. Here ASN stands for L-asparaginase (EC.3.5.1.1). By the comparison of the ASN activity expressed in the transformants, we found that the p43 promoter was the strongest promoter. Therefore, it was selected for our project. | ||
| + | |||
| + | === Experiment Method === | ||
| + | |||
| + | |||
| + | #PCR amplification of ASN gene product | ||
| + | #Construction of transformation vectors (extraction of plasmids, digestion, conjugation and transformation, etc.) | ||
| + | #Construction of Bacillus subtilis transformants | ||
| + | #SDS-PAGE electrophoresis to confirm the ASN protein expression. | ||
| + | #The enzymatic activity of ASN was determined by colorimetric method. The detection process was divided into two steps: hydrolysis and coloration of ASN. | ||
| + | #*'''ASN hydrolysis''': 100 ml diluted solution with ASN was added to the 1100 ml mixture of substrate and buffer. The reaction lasted for 10 min at 37°C. It was terminated by adding 100 ml of trichloroacetic acid (1.5 M). The mixture was then centrifuged at 12000 rpm for 2 minutes. The final system consisted of 900μL KH2PO4-K2HPO4 buffer (20 mM, pH 7.5) and 200 μl L-asparagine (189 mM). | ||
| + | #*'''Coloration''': 100ml solution from ASN hydrolysis process was added to 3400μl deionized water, and 500μl Nessler's reagent was added for coloration, the absorbance value was detected at 436 nm. (Here the definition of ASN Enzyme Activity Unit is: The amount of enzymes required to hydrolyze L-asparagine to release 1μM NH3 in 1 minute at 37°C.<br> | ||
| + | <html> | ||
| + | <center> | ||
| + | <figure> | ||
| + | <img width="40%" src="https://2019.igem.org/wiki/images/9/9a/T--BSC_United--PartI_map.png"> | ||
| + | </figure> | ||
| + | <h5>Figure 2. SDS-PAGE Electrophoresis</h5> | ||
| + | </center> | ||
| + | </html> | ||
| + | <br/> | ||
| + | |||
| + | <p>ASN enzymatic activities with different transformants:</p> | ||
| + | |||
| + | <html> | ||
| + | <center> | ||
| + | <figure> | ||
| + | <img width="70%" src="https://2019.igem.org/wiki/images/f/f4/T--BSC_United--PartIResults.png"> | ||
| + | </figure> | ||
| + | <h5>Figure 3. ANS enzymatic activity vs strain</h5> | ||
| + | </center> | ||
| + | </html> | ||
| + | <br/> | ||
| + | |||
| + | === Conclusion === | ||
| + | SDS-PAGE electrophoresis (Figure 2) and the ASN enzymatic activity comparison (Figure 3) show that the ASN production may be enhanced by the replacement of promoters. and among the promoters we have characterized, P43 has the highest start-up strength (the higher the enzyme activity, the thicker and brighter the corresponding band. | ||
<br> | <br> | ||
| − | <br> | + | We have thus characterized the basic biobricks Groe, SecA and P43 |
| − | <br> | + | |
| + | SDS-PAGE electrophoresis results: (band for the ASN protein at 43kDa is highlighted in red color) | ||
| + | Column M is the marker, column 1, 2, 3, 4, 5 and 6 are for the Bacillus subtilis constructs with the plasmids of WB0911H-ASN(original plasmid)、WBGroE、WBYxiE、WBSecA、WBYlbP and WB43, respectively. | ||
| + | |||
| + | |||
| + | ''' | ||
| + | === Characterization and measurement of P43 BioBrick:=== | ||
| + | |||
| + | Our team of Jiangnan-China 2019 described the role of P43 gene promoter and its value in part [[Part:BBa_K143013]]. | ||
| + | Quantitative measurement of P43 promoter strength: | ||
| + | This experiment tested the efficiency of P43 gene promoter. We designed a plasmid ([[Part:BBa_K2985150]]) containing P43 and pul for the quantitative determination of P43 promoter. In the plasmid, we also added a new part, which can be used by other teams for characterization and other work, details [[Part:BBa_K2985009]]. | ||
| + | |||
| + | We also measured the control characterization of sacB promoter, [[Part:BBa_K322921]]. | ||
| + | |||
| + | P43 strong promoter is a component promoter of Bacillus subtilis, which can still maintain high promoter activity under different growth conditions of colonies. We added the pullulanase gene after P43 promoter, and tested the activity of the enzyme after the plasmid was transferred into Bacillus subtilis. The strength of P43 promoter was characterized by the activity of extracellular enzyme. | ||
| + | |||
| + | We designed this unique plasmid, and then we will carry out the measurement work. We named it pma0911- P43-pul. Because the P43 promoter sequence is short, only the 56 BP sequence can not work well, so we added an auxiliary sequence before the promoter to ensure the normal operation of P43, so the P43 working sequence is 637 BP in total. | ||
| + | |||
| + | We send the sequence information to the biotechnology company and synthesize it. | ||
| + | |||
| + | The P43 promoter was connected to the plasmid by the design of the enzyme site, and the positive clone was identified by DNA electrophoresis. | ||
| + | |||
| + | [[File:Jiangnan-China P43-figure10.jpeg]]<br> | ||
| + | '''''Figure 1 P43 promoter (56bp) + pullulanase gene (bba_k2985009) + screen''''' | ||
| + | |||
| + | |||
| + | [[File:Jiangnan-China P43-figure 2.jpeg]]<br> | ||
| + | Figure 2 The positive clone DNA was identified by 0.8% agarose gel electrophoresis and PCR. The product of plasmid has a signal band at 637 BP, which is consistent with the target length. | ||
| + | |||
| + | === SDS-page detection qualitative analysis === | ||
| + | === Methods and results === | ||
| + | Take 1 mL fermented liquid, in the 12000r∙min-1, 1 min the centrifugal, the fermented supernatant as samples to identify the Protein SDS-page electrophoresis of pullulan enzyme expression, using SDS-PAGE of concentrated gum concentration was 5%, the concentration of separation gel was 9%, take samples of 30 mu, 4 x L SDS-page Protein Loading Buffer 10 μLand blending boil after 5 min, 5000 r·min-1, 2 min centrifugal, samples from 15 μL processed Protein to join the fibrin glue lane, 5 μl high molecular weight protein Marker was added. | ||
| + | |||
| + | [[File:Jiangnan-China P43-figure 3.jpeg]]<br> | ||
| + | Figure 3:An additional protein band was added near 100 kDa and was in line with the expected molecular weight of pullulanase. | ||
| + | |||
| + | After the completion of the unique plasmid construction, we introduced the plasmid into Bacillus subtilis for strain construction and screening. In addition to the screening of antibiotics, we also did plate screening, adding 2% red Pullulan in the basic LB medium, and obtained the red medium for identification. In the medium of the recombinant strain, we can get the different phenomenon that the pullulanase can and can not express. | ||
| + | |||
| + | [[File:Jiangnan-China P43-figure 4.jpeg]]<br> | ||
| + | Figure 3 medium without expression of pullulanase | ||
| + | |||
| + | [[File:Jiangnan-China P43-figure 5.jpeg]]<br> | ||
| + | Figure 4 culture medium with pullulanase expression | ||
| + | |||
| + | After the constructed recombinant strain was separated by plate scribing, the single colony was selected and cultured in 5ml LB liquid medium, and then it was vibrated at 37 ℃ and 200R·min-1 for overnight. The seed liquid was transferred to 100 ml fermentation medium with 5% inoculum amount, and cultured at 37℃, 250r·min−1. When the OD600 reached 0.6-0.8, sucrose with final concentration of 1%, 2%, 3%, 4% and 5% was added to induce the expression of pullulanase for 60h at 30℃ and 200 R·min−1. SDS-PAGE was used to detect the expression of extracellular pullulanase in the supernatant and its extracellular enzyme activity. For the relationship between extracellular enzyme activity and OD600, please refer to part [[BBa_K2985009]]. | ||
| + | |||
| + | The enzyme activity of the recombinant strain pma0911-p43-pul also reached 24.5 ± 0.3 U·ml−1. At the same time, we compared p43 with the original PHpall promoter to get the enzyme activity data. | ||
| + | |||
| + | [[File:Jiangnan-China P43-figure 6.png|600px|thumb|left|alt text]] | ||
| + | |||
| + | |||
| + | [[File:Jiangnan-China P43-figure 7.png|600px|thumb|left|alt text]] | ||
| + | |||
<!-- Add more about the biology of this part here | <!-- Add more about the biology of this part here | ||
| + | |||
===Usage and Biology=== | ===Usage and Biology=== | ||
<!-- --> | <!-- --> | ||
| − | <span class='h3bb' | + | <span class='h3bb'>Sequence and Features</span> |
<partinfo>BBa_K143013 SequenceAndFeatures</partinfo> | <partinfo>BBa_K143013 SequenceAndFeatures</partinfo> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
<!-- Uncomment this to enable Functional Parameter display | <!-- Uncomment this to enable Functional Parameter display | ||
Latest revision as of 12:47, 12 October 2022
Promoter 43 a constitutive promoter for B. subtilis
Promoter 43: Constitutive Promoter for B. subtilis
Promoter 43 is a constitutive promoter that constitutively expresses the P43 protein in B.subtilis. This promoter has been shown to be recognized and active during the exponential and lag phases of growth. It has been hypothesized that the ability to recognize the promoter in exponential and lag phase of growth is due to the recognition of the promoter by both sigma factor 55 (the major sigma factor A) and sigma factor 37 (the lag phase sigma factor B) 1. The P43 promoter has been previously used for constitutive expression of exogenous genes within B.subtilis vectors 2. The context with which we used the promoter P43 is as a Polymerase Per Second (PoPS) generator.
KEYSTONE 2022 Characterization
Promoter 43 was utilized with the gene coded for the over-expression of degQ, which has been knocked into the B. subtilis to produce the cyclolipopeptide fengycin.
Fengycins production in Bacillus subtilis 168
The main composite of our product, fengycins, is produced by engineering Bacillus subtilis 168. As it is shown in the demonstration in description, sfp and degQ genes are critical to the biosynthesis of fengycins. Besides, Bacillus subtilis 168 possesses a natural but invalid sfp gene, and the functional degQ gene is silenced with the regulation of a weak promoter.Thus, with the help of pJOE8999_sfp_degQ plasmid (Figure 1a), we knocked in both sfp gene and degQ gene in the region of the natural sfp gene of Bacillus subtilis 168 after being knocking out. Under the guidance of protocol, we successfully transformed and induced the plasmid to function as a gene editor (Figure 1c). After induction, 7 strains were selected for PCR and electrophoresis verification. Since successful knock-in would would result in a 252 bps increase in the genome of Bacillus subtilis 168, the electrophoresis result conveys preliminarily that all of these strains has achieved our target of constructing the fengycins producers (Figure 1b).

After sequencing further verified that the successful construction of the fengycins producing strain, we used the strain for fermentation (Figure 2a). For the quantification of fengycins production, the methanol - extract obtained from the fermentation broth was analyzed by high performance liquid chromatography (HPLC). The peak of fengycins appeared at the retention time of 8-11.5 min under methanol mobile phase (Figure 2c). The injection sample was a solution of lipopeptide extract from 30 mL of fermentation broth dissolved in 7 mL methanol. Besides, various concentrations of fengycins standard is also injected for the construction of the standard curve of peak area obtained by HPLC (y) and its corresponding fengycins’ concentration (x). Based on the standard curve, we got the regression curve and its corresponding formula y=9.767x+193.44, and R2>0.98 displays that the regression equation fits the observed values (Figure 2b). Substituting y=1242.3 obtained from the sample into the regression equation, the concentration of fengycin in methanol solution can be calculated as 107.4 mg/L, that is, 25.06 mg/L of fengycins can be achieved by shaking-flask fermentation.

BSC_United's Characterization to Promoter 43
Promoter P43 is a strong promoter of Bacillus subtilis expression system. It is derived from cytidine deaminase (cdd) gene in B. subtilis . It is often used to express different target proteins. Several promoters with high expression level that are commonly used in Bacillus subtilis were screened to determine the most efficient ones for our experiments on the construction of biological parts. Three of such promoters, Groe, SecA, P43, exist in the iGEM registry already. The primer sequences for individual promoters used for our synthetic biology project are as follows:
groe-up CGCGGATCCCCAATACTGTTTTCTCAAATGGTATGTA
groe-down CGGGGTACCTGAAATAACCTCCTCAATAGTATGA
YxiE-up CGCGGATCCAATTGAAGCGCGCGAAGCCA
YxiE-down CGGGGTACCGCTCTTCCCGCCTTTCGGACTG
SecA-up CGCGGATCCGGACATCGTCCGTCAGAAACGCTTT
SecA-down CGGGGTACCTCACACGCCTATTTTAGAGGCATGT
Ylbp-up CGCGGATCCGTCACAACAGTCACGTCGTGATAAA
Ylbp-down CGGGGTACCACATATATTGTAAACGCTTTATTTA
P43-up CGCGGATCCTGATAGGTGGTATGTTTTCG
P43-down CGGGGTACCTATAATGGTACCGCTATCACT
Strains and Plasmids
The bacterium strain we used was B. subtilis WB600, and the plasmid was WB0911H-ASN (with the antibiotic ampicillin and kanamycin resistance cassette). Both the strain and plasmid were kindly donated to us by Professor Jian CHEN at Jiannan University, China.
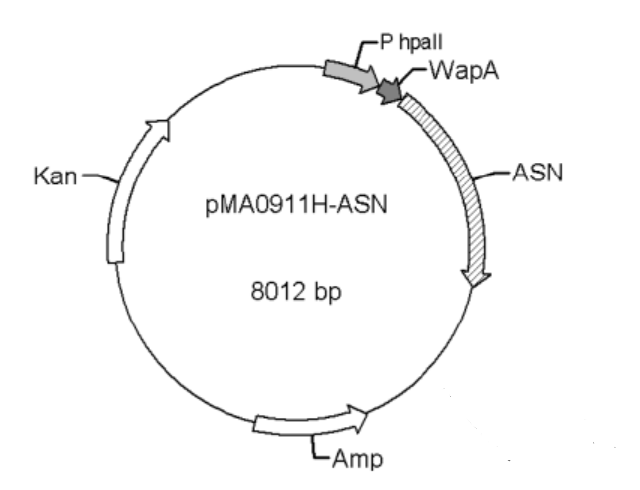
Figure 1. pMAA0911H-ASN transformation vector
The promoters Groe (BBa J100034), SecA (BBa K1469002), and P43 (BBa K143013) were used to replace the original promoter hpall in WB0911H-ASN respectively. Here ASN stands for L-asparaginase (EC.3.5.1.1). By the comparison of the ASN activity expressed in the transformants, we found that the p43 promoter was the strongest promoter. Therefore, it was selected for our project.
The promoters Groe (BBa J100034), SecA (BBa K1469002), and P43 (BBa K143013) were used to replace the original promoter hpall in WB0911H-ASN respectively. Here ASN stands for L-asparaginase (EC.3.5.1.1). By the comparison of the ASN activity expressed in the transformants, we found that the p43 promoter was the strongest promoter. Therefore, it was selected for our project.
Experiment Method
- PCR amplification of ASN gene product
- Construction of transformation vectors (extraction of plasmids, digestion, conjugation and transformation, etc.)
- Construction of Bacillus subtilis transformants
- SDS-PAGE electrophoresis to confirm the ASN protein expression.
- The enzymatic activity of ASN was determined by colorimetric method. The detection process was divided into two steps: hydrolysis and coloration of ASN.
- ASN hydrolysis: 100 ml diluted solution with ASN was added to the 1100 ml mixture of substrate and buffer. The reaction lasted for 10 min at 37°C. It was terminated by adding 100 ml of trichloroacetic acid (1.5 M). The mixture was then centrifuged at 12000 rpm for 2 minutes. The final system consisted of 900μL KH2PO4-K2HPO4 buffer (20 mM, pH 7.5) and 200 μl L-asparagine (189 mM).
- Coloration: 100ml solution from ASN hydrolysis process was added to 3400μl deionized water, and 500μl Nessler's reagent was added for coloration, the absorbance value was detected at 436 nm. (Here the definition of ASN Enzyme Activity Unit is: The amount of enzymes required to hydrolyze L-asparagine to release 1μM NH3 in 1 minute at 37°C.
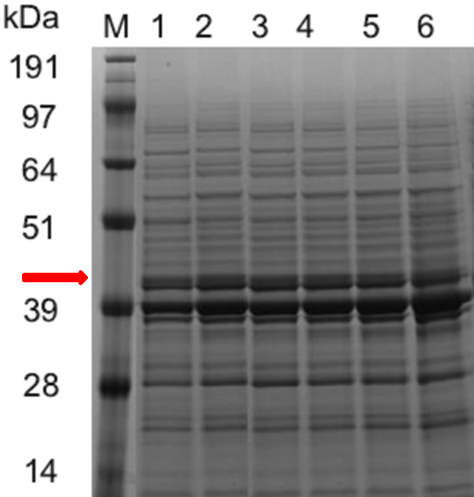
Figure 2. SDS-PAGE Electrophoresis
ASN enzymatic activities with different transformants:
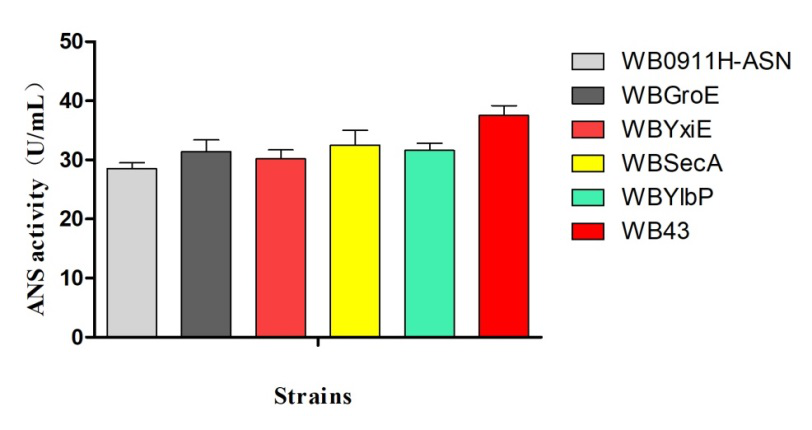
Figure 3. ANS enzymatic activity vs strain
Conclusion
SDS-PAGE electrophoresis (Figure 2) and the ASN enzymatic activity comparison (Figure 3) show that the ASN production may be enhanced by the replacement of promoters. and among the promoters we have characterized, P43 has the highest start-up strength (the higher the enzyme activity, the thicker and brighter the corresponding band.
We have thus characterized the basic biobricks Groe, SecA and P43
SDS-PAGE electrophoresis results: (band for the ASN protein at 43kDa is highlighted in red color) Column M is the marker, column 1, 2, 3, 4, 5 and 6 are for the Bacillus subtilis constructs with the plasmids of WB0911H-ASN(original plasmid)、WBGroE、WBYxiE、WBSecA、WBYlbP and WB43, respectively.
Characterization and measurement of P43 BioBrick:
Our team of Jiangnan-China 2019 described the role of P43 gene promoter and its value in part Part:BBa_K143013. Quantitative measurement of P43 promoter strength: This experiment tested the efficiency of P43 gene promoter. We designed a plasmid (Part:BBa_K2985150) containing P43 and pul for the quantitative determination of P43 promoter. In the plasmid, we also added a new part, which can be used by other teams for characterization and other work, details Part:BBa_K2985009.
We also measured the control characterization of sacB promoter, Part:BBa_K322921.
P43 strong promoter is a component promoter of Bacillus subtilis, which can still maintain high promoter activity under different growth conditions of colonies. We added the pullulanase gene after P43 promoter, and tested the activity of the enzyme after the plasmid was transferred into Bacillus subtilis. The strength of P43 promoter was characterized by the activity of extracellular enzyme.
We designed this unique plasmid, and then we will carry out the measurement work. We named it pma0911- P43-pul. Because the P43 promoter sequence is short, only the 56 BP sequence can not work well, so we added an auxiliary sequence before the promoter to ensure the normal operation of P43, so the P43 working sequence is 637 BP in total.
We send the sequence information to the biotechnology company and synthesize it.
The P43 promoter was connected to the plasmid by the design of the enzyme site, and the positive clone was identified by DNA electrophoresis.

Figure 1 P43 promoter (56bp) + pullulanase gene (bba_k2985009) + screen

Figure 2 The positive clone DNA was identified by 0.8% agarose gel electrophoresis and PCR. The product of plasmid has a signal band at 637 BP, which is consistent with the target length.
SDS-page detection qualitative analysis
Methods and results
Take 1 mL fermented liquid, in the 12000r∙min-1, 1 min the centrifugal, the fermented supernatant as samples to identify the Protein SDS-page electrophoresis of pullulan enzyme expression, using SDS-PAGE of concentrated gum concentration was 5%, the concentration of separation gel was 9%, take samples of 30 mu, 4 x L SDS-page Protein Loading Buffer 10 μLand blending boil after 5 min, 5000 r·min-1, 2 min centrifugal, samples from 15 μL processed Protein to join the fibrin glue lane, 5 μl high molecular weight protein Marker was added.

Figure 3:An additional protein band was added near 100 kDa and was in line with the expected molecular weight of pullulanase.
After the completion of the unique plasmid construction, we introduced the plasmid into Bacillus subtilis for strain construction and screening. In addition to the screening of antibiotics, we also did plate screening, adding 2% red Pullulan in the basic LB medium, and obtained the red medium for identification. In the medium of the recombinant strain, we can get the different phenomenon that the pullulanase can and can not express.

Figure 3 medium without expression of pullulanase

Figure 4 culture medium with pullulanase expression
After the constructed recombinant strain was separated by plate scribing, the single colony was selected and cultured in 5ml LB liquid medium, and then it was vibrated at 37 ℃ and 200R·min-1 for overnight. The seed liquid was transferred to 100 ml fermentation medium with 5% inoculum amount, and cultured at 37℃, 250r·min−1. When the OD600 reached 0.6-0.8, sucrose with final concentration of 1%, 2%, 3%, 4% and 5% was added to induce the expression of pullulanase for 60h at 30℃ and 200 R·min−1. SDS-PAGE was used to detect the expression of extracellular pullulanase in the supernatant and its extracellular enzyme activity. For the relationship between extracellular enzyme activity and OD600, please refer to part BBa_K2985009.
The enzyme activity of the recombinant strain pma0911-p43-pul also reached 24.5 ± 0.3 U·ml−1. At the same time, we compared p43 with the original PHpall promoter to get the enzyme activity data.


Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]