
Part:BBa_K3017001
CRISPRi sgRNA for gfp DNA binding - transcription template


dCas9 protein is directed to the specific DNA locus by a single-guide RNA (sgRNA), where it binds to suppress downstream gene expression. With reference to the research [1] on reversible CRISPRi switch, we redesigned the traditional sgRNA by adding an artificial linker behind crRNA and tracrRNA and modified the 3-component-sgRNA to suit our suppression purpose. Our design of sgRNA is compatible with spCas9.
Spacer - crRNA
crisprRNA(crRNA) is also commonly referred to as the spacer. When choosing the target binding region, we considered mainly 2 factors, namely the location of the PAM sequence and the suppression effect upon binding.
The research[1] shows CRISPRi suppression effect is the strongest 35nt upstream start codon of the coding region. However, the area upstream of our coding region is a generic constitutive promoter. To avoid non-specific binding, we compromised the suppression efficiency and chose a region shortly after the start codon, where suppression is only a few percents weaker than the ideal region. We found a PAM sequence (TGG) 27nt into gfp part BBa_E0040, which lead to a sgRNA binding region spanning 20bp, 7nt into CDS. To accommodate the PAM sequence in BBa_E1010 mrfp, the spacer is arranged on the opposite DNA strand, 14nt into the gene. When this part, specific to gfp is transcripted, GFP is suppressed.
Handle - tracrRNA
tracrRNA is an RNA loop that acts as a handle for dCas9 to hold onto. So that the dCas9 protein is delivered to the target site together with the sgRNA. Experiments have proved that tracrRNA is strictly required for Cas9-mediated DNA interference both in vitro and in vivo[4]. The tracrRNA forms a loop on the sgRNA after transcription to provide a scaffolding site for the dCas9 to form a duplex with the spacer.
Loop - artificial linker
Destroying the secondary structure of the handle in sgRNA could theoretically cause dissociation of the dCas9 protein from the sgRNA, thus, removing the suppression effect. The study mentioned above had proved this hypothesis correct. The team[1] then tried to design an artificial linker, which also forms a loop as a secondary structure, after the handle. After several trials and modifications, the research team discovered that extending the artificial loop, i.e. destroying the secondary structure, could further increase the derepression.
A corresponding antisense RNA (asRNA), K3017003, is responsible for reversing the repression effect on gfp induced by this sgRNA. Part of the antisense is complementary with the artificial linker of this sgRNA.
Characterizations
This part involve interaction with part BBa_K3017003 so they were being characterized as a pair. Using sgRNA-terminator BBa_K3017061 and asRNA-terminator-BBa_K3017060 as a model, we have characterized this part which is highly similar in function.
RNA In Vitro Transcription (IVT) and interaction assay
Aim:
To assess whether the sgRNA and asRNA can be transcribed successfully and hybridized in vivo, an in vitro assay was designed to assess the transcription of the RNAs and the feasibility for the RNAs to hybridize in vitro.
Flow of experiment:
sgRNA and asRNA is synthesized via in vitro transcription (NEB HiScribe T7 in vitro transcription kit). Part of the transcript is mixed together to allow for hybridization. The part is then analyzed on both formaldehyde denaturing agarose gel and non-denaturing agarose gel to assess transcript size and hybridization respectively. (figure 1)
| Denaturing Gel (formaldehyde) | Non-denaturing Gel |
| Destroys secondary structure | Preserves secondary structure |
| Assess actual size of transcript | Assess interaction |

Step 1 - PCR synthesis of IVT templates
The 2 RNA’s transcription templates, designed according to NEB IVT kit manual, were created by using PCR to insert a T7 promoter in front of the transcription template using an overhang forward primer. (Figure 2)

Thereafter, the PCR product is run through an agarose gel to separate impurities and inspect the sizes of the IVT templates before undergoing gel purification.
Results:
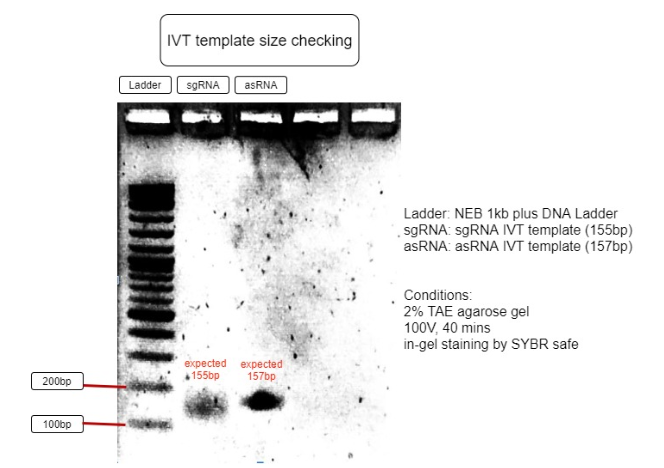
Figure 3. Verification of RNA’s transcription templates
The size of IVT template is as expected to be around 150bp, with sgRNA expected to be 155bp and asRNA expected to be 157bp. This proves the IVT template sizes are correct and can be purified for IVT reaction. (Figure 3)
Step 2 - In vitro transcription (IVT) using NEB T7 HiScribe Transcription Kit
To synthesize the sgRNA and asRNA, 75ng of gel-purified DNA transcription template was added into the IVT reaction mixture. The reaction is set for 16 hours of incubation at 37°C, using a thermal cycler to prevent evaporation.
The transcripted RNAs is treated with DNase1, then precipitated by sodium acetate/ethanol.
The sizes of the 2 precipitated RNAs are confirmed by using denaturing formaldehyde-MOPS 2% agarose gel. Since sgRNA is known to form multiple secondary structures, heat denaturation at 65 °C for 7 minutes is needed prior to loading into the gel to remove all secondary structures, allowing for accurate assessment of its size.
Results:

Figure 4. RNA transcription check
The gel photo above clearly indicates the successful transcription of the sgRNA and asRNA, with the size matching their expected size (sgRNA=138bp, asRNA=140bp).
Step 3 - RNA in vitro hybridization and visualization
In order to test the environment needed for in vitro hybridization, 4 different environments were tested in a combination of using 3x Hybridization Buffer (HB) and the use of initial denaturation. Regardless of the conditions being tested, the 2 RNAs are incubated together at 37°C for 45 minutes, before snap cooling to 0°C to preserve the hybridized structure and RNase degradation. (protocol we used for the experiment)
It is known that salt content is crucial to RNA duplex formation, thus, 3x HB is used to provide the necessary salt content and stabilize the RNAs. DEPC water is used as a negative control to the testing of hybridization buffer.
Since this assay is designed as an in vitro demonstration of the in vivo hybridization, the temperature of incubation is set to 37°C to simulate the in vivo environment with no initial denaturation. As a fail-safe test, the necessity for initial denaturation is also tested as the in vitro hybridization is expected to be less efficient without the Hfq Chaperone. Therefore, denaturing the RNAs before slow cooling will remove the RNAs secondary structure while promoting the complementation and hybridization during slow cooling.
A non-denaturing 2% MOPS agarose gel is then used to visualize the hybridization.
Results:
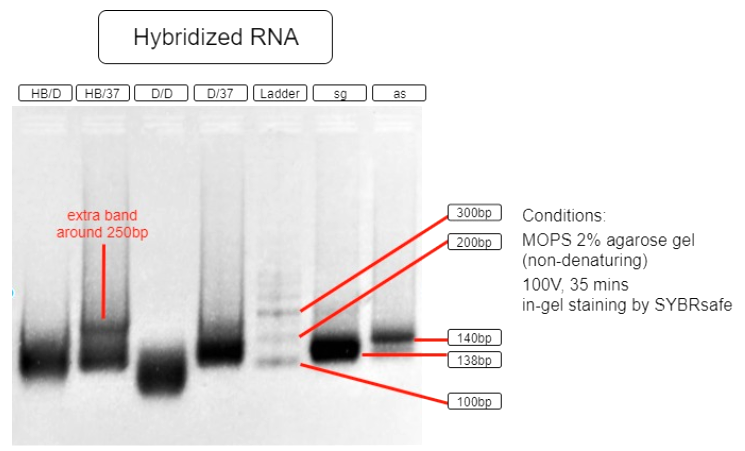
Figure 5. RNA hybridization assay
| HB/D | Hybridization Buffer + denature + slow cool* + 37°C incubation |
| HB/37 | Hybridization Buffer + slow cool* + 37°C incubation |
| D/D | DEPC H2O + denature + slow cool* +37°C incubation |
| D/37 | DEPC H2O + 37°C incubation |
| L | Ambion Century™ - Plus RNA Markers |
| sg | sgRNA BBa_K3017061 (138bp) |
| as | asRNA BBa_K3017060 (140bp) |
*slow cool is done by cooling the RNA sample using thermo cycler at 5°C per 10 sec until incubation temperature
As shown in the above gel photo (Figure 5), only lane AB/37 shows an extra band which appears to be around 250bp when compared to the ladder. This band indicates the hybridization of sgRNA and asRNA and is therefore running slower in agarose gel.
This result shows that salt content provided by the hybridization buffer (60mM KCl, 6mM HEPES, 0.2mM MgCl2 at 1x), which is similar to that of a cellular salinity, is crucial to the hybridization. This result also indicates that the RNAs are able to hybridize with each other in 37°C incubation with no initial denaturation needed. These results suggest that the RNAs are capable of hybridizing in vivo. With endogenous expression of Hfq protein in E.coli, the asRNA will also stabilize due to Spot 42 and attract other ssRNAs to hybridize.
Thus, we suspect the in vivo hybridization efficiency would be higher than our results.
References:
[1] Y. J. Lee, A. Hoynes-Oconnor, M. C. Leong, and T. S. Moon, “Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system,” Nucleic Acids Research, vol. 44, no. 5, pp. 2462–2473, Feb. 2016.
[2] C. Anders, O. Niewoehner, A. Duerst, and M. Jinek, “Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease,” Nature, vol. 513, no. 7519, pp. 569–573, 2014.
[3] S. H. Sternberg, S. Redding, M. Jinek, E. C. Greene, and J. A. Doudna, “DNA Interrogation by the CRISPR RNA-Guided Endonuclease Cas9,” Biophysical Journal, vol. 106, no. 2, 2014.
[4] T. Karvelis, G. Gasiunas, A. Miksys, R. Barrangou, P. Horvath, and V. Siksnys, “crRNA and tracrRNA guide Cas9-mediated DNA interference inStreptococcus thermophilus,” RNA Biology, vol. 10, no. 5, pp. 841–851, 2013.
[5] T. Møller, T. Franch, P. Højrup, D. R. Keene, H. P. Bächinger, R. G. Brennan, and P. Valentin-Hansen, “Hfq,” Molecular Cell, vol. 9, no. 1, pp. 23–30, 2002.
[6] G. M. Cech, A. Szalewska-Pałasz, K. Kubiak, A. Malabirade, W. Grange, V. Arluison, and G. Węgrzyn, “The Escherichia Coli Hfq Protein: An Unattended DNA-Transactions Regulator,” Frontiers in Molecular Biosciences, vol. 3, 2016.
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
| None |