
Part:BBa_K1989002
Triplespytag with mSA and His-tag
Usage and Biology
In the last few years, hydrogens made from natural or synthetic polymers have been investigated due to their extensive application in clinical medicine and synthetic biology. Compared to traditional biological material, protein-based multifunctional biological material is low-cost, facile and eco-friendly. However, strategies for assembling 3D molecular networks synthesized only by protein molecular remain underdeveloped. The reason why investigating this technology is still tough is lack of protein-based cross linking agents. Inspired by the self-catalysis of isopeptide bond between Lys and Asp in Streptococcus pyogenes fibronectin-binding protein FbaB, researchers split the catalytic domain and obtained two peptide called SpyTag(the short one) and SpyCatcher(the long one) which are able to form isopeptide bond with the other without any assistant. By fusing SpyTag and SpyCatcher with functional domains respectively, researchers solved the problem tactfully. In order to using SpyTag and SpyCatcher system as scaffold, we fused three SpyTag spaced by (VPGVG)4 with 6xHistag in N-terminal and another functional protein called monomeric streptavidin(mSA) in C-terminal.
Based on our results, the fused protein His-3A-mSA (3A-SUP) possess both isopeptide bond forming function and biotin-binding ability. Thus, using 3A-mSA and biotinylated magnetic bead as parts of hydrogel formation, we can recycle our multifunctional biomaterial.
You can find triplespytag with SUP and His-tag here https://parts.igem.org/Part:BBa_K1989000", and Triplespycatcher with His-tag here https://parts.igem.org/Part:BBa_K1989001".
Cultivation, Purification and SDS-PAGE
Cultivation
The part was assembled with T7 promoter and RBS in pET28a plasmid vector. E. coli strain BL21(DE3) harboring the appropriate plasmid was grown at 37 °C in 2xYT medium overnight with suitable concentration of antibiotic. The culture was diluted 100 fold into fresh medium with antibiotic and grown at 37°C to an optical density of 0.6~0.8 at 600 nm, the protein expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and cells were grown overnight at 25°C.
Purification
Cells were centrifuged at 8000rpm for 15min at 4°C. Resuspend the cell paste expressing recombinant protein in binding buffer (20 mM Tris-HCl, 0.5 M NaCl, 20 mM imidazole, 1mM β-mercaptoethanol, pH7.4), containing SIGMAFAST™ Protease Inhibitor Cocktail Tablets (SIGMA-ALORICH). Disrupt the cells with sonication for 20 min with suitable power on ice and centrifuge at 18000 rpm for 40 min at 4°C. Remove remaining particles by passing the supernatant through a 0.22 μm filter. The HisTrap™ column (GE Healthcare, Inc.) was equilibrated with binding buffer. Load the sample and wash the column with binding buffer.
Elute the target protein with a linear gradient starting with binding buffer and ending with the same buffer including 500mM imidazole. The eluted fraction containing the target protein were concentrated by Amicon® Ultra Centrifugal Filters (Merck) with a 10 kDa cutoff, then frozen by liquid nitrogen and stored at -80°C.
SDS-PAGE
Protein purification was checked by SDS-PAGE and the resulting protein is quantified by Braford analysis.
Activity Analysis
Gel Formation
Basic Exploration
Exploring the polymerization of our crosslinking network is significant to subsequent application. We must know what our Spy Network is like, and how long they need to complete their reactions, which would offer us a kind of intuition of their crosslinking ability.
To answer the questions shown above, we have analyzed the crosslinking ability of our fundamental monomers, 3A, 3A-SUP, 3A-mSA and 3B. They were firstly diluted to certain concentration, and were made to react under the restriction that the number of A was equal to B. The monomers containing A were conducted to react with 3B at 25℃ and pH=7.3 with the duration of 2 hours.

Fig. 1. Exploration of the polymerization ability of the 3A-SUP/3A-mSA with 3B. Left: the Coomassie Blue staining gel of basic experiment, which illustrates the basic cross-linking ability of 3A-SUP/3A-mSA and 3B. Lane 1, Thermo® Protein Marker; lane 2, 3A-SUP; lane 3, 3A-mSA; lane 4, 3A-SUP+3A-mSA; lane5, 3B; lane 6, 3A-SUP+3B; lane 7, 3A-mSA+3B. (Molecular Weight: 3A-SUP, 21.4kDa; 3A-mSA, 25.4kDa; 3B, 55.4(62) kDa. Right: A, the OD value of the lane 5 of oligomers produced by the mix of 3A-SUP and 3B. Peaks illustrate the monomers and the possible products, ①, 3A-SUP (21.4kDa); ②, 3B (62kDa); ③, 1x 3A-SUP+1x 3B (83.4kDa); ④, 3x 3A-SUP+1x 3B (126.2kDa); ⑤, 2x 3A-SUP+2x 3B (166.8kDa); ⑥, 4x 3A-SUP+4x 3B (333.6kDa). B, the OD value of the lane 6 of oligomers produced by the mix of 3A-mSA and 3B. Peaks illustrate the monomers and the possible products, ①, 3A-mSA (25.4kDa); ②, 3B (62kDa); ③, 1x 3A-mSA+1x 3B (87.4kDa); ④, 1x 3A-mSA+2x 3B (149.4kDa); ⑤, 2x 3A-mSA+2x 3B (174.4kDa); ⑥, 3x 3A-mSA+4x 3B (324.2kDa). (The software lane 1D was used to draw the graph.) (“3A-SUP” stands for “Triple SpyTag-SUP”, “3A-mSA” for “Triple SpyTag-mSA”, and “3B” is the abbreviation of “Triple SpyCatcher”)
We found that some new bands appeared above the band of 3B when it was mixed with monomers containing A, which demonstrated that our idea of forming functional hydrogel was executable. The products were mainly oligomers (Fig. 2 A-F, Table 1), for it is easy to form loops, which hindered the linkage between different monomers at such low concentration. Interestingly, with the restriction that A is equal to B in number, and the content of 3B was constant initially, the crosslinking ability at low concentration of these monomers were different with each other by comparing the surplus content of 3B. What’s more, it was surprising that the position of 3B band (~62kDa) was not accord with its theoretical weight (55.4kDa). Further experiments will be done to understand the differences.
Gradient Experiment
After exploring the basic character, i.e., the ability to crosslink, of our reactants, we would like to further explore the advantageous conditions for our polymerization, such as pH gradient, temperature gradient, and concentration gradient of monomers.
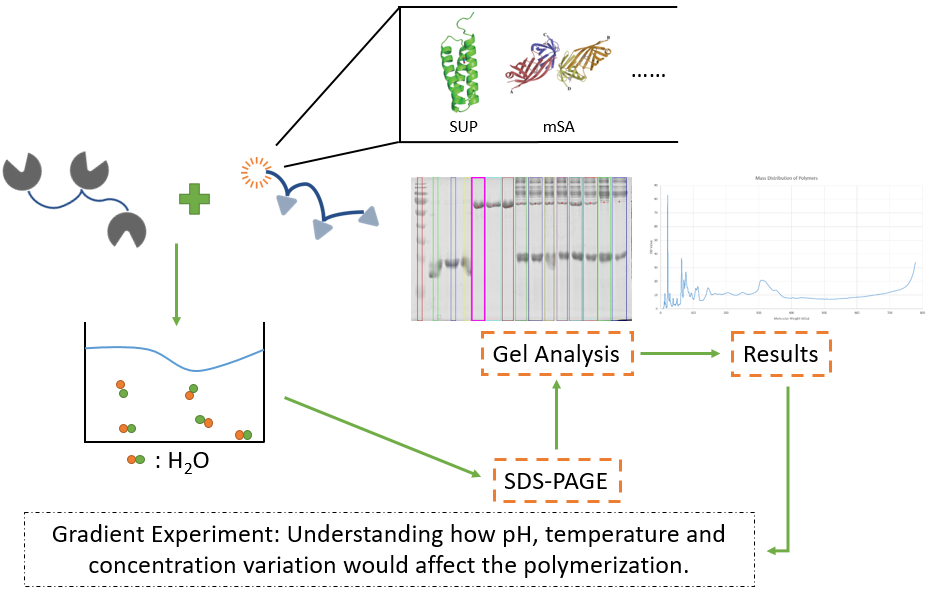

Fig. 2. Design the gradient experiment of pH, temperature and concentration
First experiment is concerned with pH gradient. Given that our Spy Network in practical application would face water environment with pH nearly neutral, and our proteins have their own optimum pH of functioning, we decide to make pH gradient of 6.3/7.3/8.3. Typical monomers 3A-SUP and 3B would firstly dissolve in TBS at certain pH values, then they were put to react at mentioned pH gradient. The reaction would be conducted at 25℃, with duration of 2 hours. Samples would be extracted every 30min to make clear the variation tendency of monomers. After the reaction, SDS-PAGE would be conducted and the gels would be scanned and analyzed by the software Lane 1D. Thus the relationship between time and the content of 3B and 3A would be found out. Three parallel groups were set to realize quantitative analysis.

Fig. 3. pH Gradient Experiment A: the reactive extent for 3B at different pH. Experiments were repeated three times and error bars were added. B: The mass distribution of oligomers at different time at pH7.3 (70kDa-450kDa).
From the results of analyzing SDS-PAGE gels showing above, we clearly observed the changing law of 3B and the mass distribution of polymer homologs. Appropriately lower pH would lead to faster consumption of monomers, while our time scale should be smaller in order to show the condensation polymerization nature.
Second is concerned with temperature. According to the common environmental temperature, three typical values, 16℃, 25℃, 37℃ were introduced in our temperature experiment. Similar to pH Gradient Experiment, typical monomers 3A-SUP and 3B would firstly dissolve in TBS at pH=7.3, then they were put to react at mentioned temperature gradient. The reaction would be conducted at pH=7.3, with the duration of 2 hours. The subsequent steps were same to that of pH Gradient Experiment.

Fig. 4. Temperature Gradient Experiment A: the reactive extent for 3B at different temperature. Experiments were repeated three times and error bars were added. B: The mass distribution of oligomers at different time at 16℃ (70kDa-400kDa).
The analysis of SDS-PAGE gel clearly showed the variation tendency of the surplus content of 3B as a function of time, and of the mass distribution of homologs. According to the result, we could conclude that higher temperature would lead to the faster consumption of monomer, and at designated time scale we didn’t see any change in the change of weight distribution of polymer homologs. We supposed that it should be easier to form loops (intramolecular reactions) at such low concentration, and the time scale is too large to show the variation. Though the rate of polymerization should be described by the consumption rate of functional groups, we decide to use the consumption rate of monomers to simply describe the tendency.
Third is concerned with concentration. At low concentration would cross-linkage not polymerize, but oligomerize (The difference between these two reactions can be described by crosslinking density, the former is large, while the latter is rather small.), according to the basic knowledge of Polymer Science. From this point of view, if we hope to promote the mass, strength and contact area of polymers, we have to mix our monomers at high concentration. Thus, understand the relationship between the concentration and the variation of mass distribution of polymers is quite essential.

Fig. 5. The mass distribution of oligomers of concentration gradient experiment at different concentration Left: the Coomassie Blue staining gel of concentration gradient experiment, which clearly showed the change of mass distribution according to the change of concentration. Lane 1, Thermo® Protein Marker; lane 2, 3A-SUP; lane 3, 3A-mSA; lane 4, 3A-SUP+3A-mSA; lane5, 3B; (1mg/mL) lane 6, 3A-SUP+3B; lane 7, 3A-mSA+3B; lane 8, 3A-SUP+3A-mSA+3B; (5mg/mL) lane 9, 3A-SUP+3B; lane 10, 3A-mSA+3B; lane 11, 3A-SUP+3A-mSA+3B; (10mg/mL) lane 12, 3A-SUP+3B; lane 13, 3A-SUP+3A-mSA(5mg/mL)+3B. (Molecular Weight: 3A-SUP, 21.4kDa; 3A-mSA, 25.4kDa; 3B, 55.4(62)kDa. “3A-SUP” stands for “Triple SpyTag-SUP”, “3A-mSA” for “Triple SpyTag-mSA”, and “3B” is the abbreviation of “Triple SpyCatcher”). 5mg/mL and 10mg/mL samples were diluted to 1mg/mL after the reaction (with the duration of 2 hours). The mass of samples in lanes was equal to each other. Right: the OD value of the lanes 2, 5, 6, 9, 12 of the oligomers produced by the mix of 3A-SUP and 3B. Peaks illustrate the monomers and the possible products, ①, 3A-SUP (21.4kDa); ②, 3B (62kDa); ③, 1x 3A-SUP+1x 3B (83.4kDa); ④, 3x 3A-SUP+1x 3B (126.2kDa); ⑤, 2x 3A-SUP+2x 3B (166.8kDa); ⑥, 4x 3A-SUP+4x 3B (333.6kDa); ⑦, average mass of polymers larger than 750kDa (~6x 3A-sup+10x 3B or 9x 3A-SUP+9x 3B). (The software lane 1D was used to draw the graph.)
The analysis of SDS-PAGE gel showed that more hyper-branched products with heavier molecular weight appeared with the increase of concentration, which, could also lead to the prediction that the average weight would be larger if higher concentration of monomers were mixed together.
Biotin-binding ability
To make our material retrievable, a recombinant protein consisting of mSA and 3A was designed. So that the mSA module could participate as triple SpyTag-mSA in the formation of the hydrogel. We ligated biotin molecules to amino-coated magnetic beads. When we use magnet to immobilize these beads, the protein hydrogel full of uranyl could be retrieved.

Fig. 6. Design of the retrieval
It was important for us to know if it worked well of the fusion protein--- triple SpyTag- mSA. We mixed 4μL biotin-coated magnetic beads (10mg/ml) or recycled beads (which were boiled and washed by with PBST containing 1M NaCl for 3 times) with 40μL triple SpyTag or triple SpyTag- mSA (1mg/ml). Incubated the mixture at 37℃ for 1 hour with 1500 rpm in a shaker. Vibration was necessary to avoid the aggregation of beads. After incubation, we took 10μL sample for SDS-PAGE analysis, and another 10 μL for Bradford assay. These two assays aimed to see the results of collection.

Fig. 7. Statistical analysis of binding efficiency of mSA. ****p < 0.0001. **p < 0.01. n=3. Error bars indicate SD.
The SDS-PAGE showed 3A-mSA could be adsorbed efficiently but 3A couldn’t. Bradford assay measured the residual proteins in the environment quantificationally. Only 14.09% 3A-mSA remained after treating with biotin-coated magnetic beads for an hour. If the beads were boiled and washed, the binding efficiency would reduce and 27.27% 3A-mSA would be remained. In the meanwhile, if the protein didn’t contain mSA module such as triple SpyTag, the binding efficiency would be quite low, and there was 78.44% (using the new beads) or 91.45% (using recycled beads) 3A remained.
These qualitative and quantificational results proved the protocol we used above were functional, and our biotin-coated beads could exhibit excellent absorption capacity to combine with mSA module as well as high specificity. It’s also shown that the beads remain valid after boiled and washed, with about only 15% absorption capacity lost. What's more, it was proved that our biotin-coated beads could be recycled for once or more to reduce costs and get more environment-friendly.
We had proved 3A-mSA could have normal function. But we still didn’t know if the hydrogel contained mSA module could be retrieved. So we mixed three kinds of monomers together and waited for 1 hour to let them crosslink as far as possible. The Sample list was shown in Fig.4. To evaluate the biotin binding efficiency of Spy network, the hydrogel was incubated with biotin-coated magnetic beads at 37℃ for 1 hour with 1500 rpm in a shaker.

Fig. 8. Sample list for evaluating the efficiency of retrieving protein hydrogel
The Bradford assay was used to measure the concentration of remaining protein. A remarkable decrease was shown when incubating the reaction system with beads for 1h. Only 22.13% protein component was remained. It meant that the biotin-coated beads could be combined with the mSA module even if in the hydrogel.

Fig. 9. Harvest of protein hydrogel by biotin coated beads. Statistical analysis of binding efficiency of Spy network. ***p < 0.001. n=3. Error bars indicate SD.
To assess if other protein modules in the hydrogel could be adsorbed by mSA module, we did another experiment to test the change of fluorescence intensity of mRFP module with or without beads treatment. The experimentation was the same as above. The excitation peak of mRFP was 584 nm, and the emission peak was 607 nm. The fluorescence intensity was measured by a plate reader.

Fig. 10. The remaining proportion of fluorescence intensity with beads treated. ****p < 0.0001. Error bars indicate SD.
The fluorescence intensity of control groups was about 12.83 a.u. while in the experiment groups this value was 3.19 a.u. Only 24.90% fluorescence was remained, indicating that a majority of 3A-RFP module could be assembled into the hydrogel, and with the absorption of the hydrogel by 3A-mSA module, 3A-RFP module can be taken away. It also means that, if we use 3A-SUP in this hydrogel, the biotin coated beads can also drag it away, with uranyl binding to it.
References
1. Sun, F. et al. Proc. Natl Acad. Sci. USA 111, 11269-11274 (2014).
2. DeMonte, Daniel. et al. Proteins 81, 1621-1633 (2013).
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25INCOMPATIBLE WITH RFC[25]Illegal AgeI site found at 486
Illegal AgeI site found at 546 - 1000COMPATIBLE WITH RFC[1000]
Functional Parameters
| None |