
Part:BBa_J64997
T7 consensus -10 and rest
Characterization in vivo
This part has been characterized for GFP expression in the pCDF plasmid by the 2015 LASATX iGEM team.
We placed the T7 promoter in front of the sfGFP gene (BBa_K1624004). The experimental construct was placed in a pCDFDuet-1 vector and transformed into BL21(DE3) cells. We used BL21(DE3) cells without the plasmid containing T7 as our negative control. Cells were grown overnight. A 1:100 dilution was made, and cultures were grown with 20% glucose to 0.6 OD. At 0.6 OD, cells were resuspended in IPTG and induced for 2 hours. After induction, cells were washed and resuspended in PBS buffer. Fluorescence was then measured at 600 nm for 25 flashes. Bars show average of triplicates. Error bars show standard deviation.

The sfGFP gene placed downstream of the T7 promoter was successfully expressed, with fluorescence from the experimental construct significantly higher than the control.
Characterization in Cell-Free Environment
The T7 promoter was characterized in self-made E.Coli lysate from strain BL21(DE). Cell-free T7-GFP-mut3b versus GFP-mut3b (BBa_E0040) synthesis was analyzed. Fluorescence was measured at 37°C for five hours on a plate reader. For details on how the lysate and the energy solution were made and which components went into the final reaction volume of 10uL, check out our [http://2017.igem.org/Team:EPFL/Protocols protocols]. Shown are two times three repeats and a negative control, a shaded error graph (control was subtracted) and a bar plot summarizing the result.
GFP-mut3b expression cannot be dinstinguished from the no DNA control. T7-GFP-mut3b expression however is high. Saturation occurs after about five hours. Adding the T7 promoter in front of the GFP-mut3b enables transcription by the T7 polymerase, a much more efficient polymerase than the E.Coli polymerase found natively in E.Coli cells.



Improvements
This part was used as a promoter in the following parts : BBa_K2203001, BBa_K2203002, BBa_K2203003, BBa_K2203004, BBa_K2203005, BBa_K2203006 ; BBa_K3100022 (by SCUT_China 2019, https://parts.igem.org/Part:BBa_K3100022).
This part was also used as a promoter in the following parts: BBa_K3352008, BBa_K3352009 (by TAS_Taipei 2020, https://parts.igem.org/Part:BBa_K3352008, https://parts.igem.org/Part:BBa_K3352008).
Improvement of BBa_K3100022 by SCUT_China 2019
In order to achieve a larger scale and more accurate regulation range, we have improved T7 promoters(BBa_J64997) with different strength providing more options for the precise regulation.

Fig1. T7 promoters with different strength
Improvement of BBa_K3352000 and BBa_K3352001 by TAS_Taipei 2020
We improved the characterization of BBa_J64997. The part T7 consensus -10 and rest serves as a T7 promoter that allows the binding of T7 RNA Polymerase to initiate transcription (Arnaud-Barbe, 1998). We decided to further characterize this part to provide data on whether this promoter sequence serves a benefit to the expression of enzymes compared to other promoters, specifically enzymes such as ligases, which are not only indispensable in bioanalytical techniques but also in viral diagnostic kits such as ours.
In order to characterize this existing part, we flanked a pET3a T7 promoter with downstream BBa_K3352002, BBa_K3352000, and BBa_K3352003, which forms the composite part BBa_K3352008. Since the pET3a T7 promoter has the same sequence as the T7 consensus -10 and rest, the SplintR Ligase enzyme expressed through this construct serves as an indicator of protein expression levels for this T7 promoter sequence.
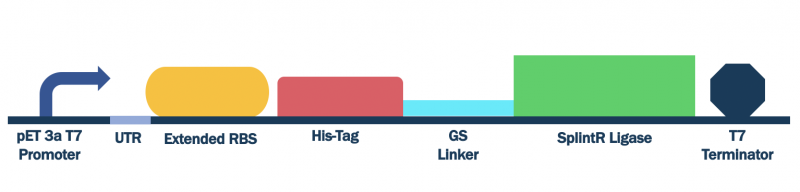
Figure #1. Design of pET Parts + SplintR Ligase Expressing Construct (BBa_K3352008).
Plasmids containing this construct was transformed into DH5⍺ E. coli cells for plasmid replication and subsequently miniprepped to obtain a high yield of plasmid. We then transformed these plasmids into BL21(DE3) E. coli cells. Growing an overnight culture and measuring the OD600 to dilute cells to standardized populations, we induced expression with 0.1M IPTG once the OD600 surpassed 0.5. Liquid cultures were grown for an additional 2 hours.
While growing our cultures, we collected culture samples both before and after IPTG induction. We subsequently ran SDS-PAGE with these samples. Our results in Figure #2 indicates that bands of the enzyme were in fact present at the 35.2kDa band, the molecular weight of our SplintR Ligase enzyme (including the 6x histidine tag and GS linker attached). However, due to the presence of other notable proteins, we seeked further validation to ensure that our enzyme of interest was expressed.
After protein expression, we harvested and lysed the cells with xTractor Lysis Buffer and centrifuged them for sample preparation (XTractorTM Buffer & XTractor Buffer Kit User Manual, n.d.). This was followed by protein purification through Ni sepharose affinity chromatography, which could isolate our his-tagged SplintR Ligase enzymes.
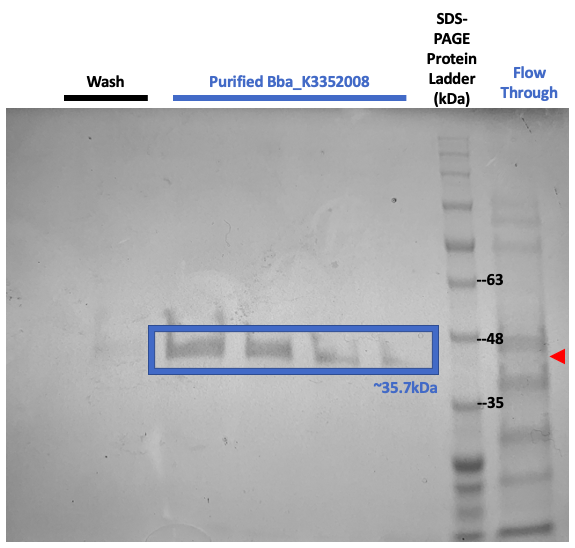
Figure #3. SDS-PAGE on purified proteins with the pET3a Parts SplintR Ligase expressing construct (BBa_K3352008).
We once again ran SDS-PAGE but with the purified samples. These results shown in Figure #3 once again suggests that our now purified enzyme of interest, SplintR Ligase, was produced through the expression of our construct design as seen through the band at 35.2kDa.
To compare the protein expression capability of this T7 promoter, we conducted the same test but replaced our T7 promoter from our previously used composite part BBa_K3352008 with a strong constitutive promoter BBa_J23100. This formed our construct BBa_K3352004, whose SplintR Ligase expression can be compared to that of BBa_K3352008.
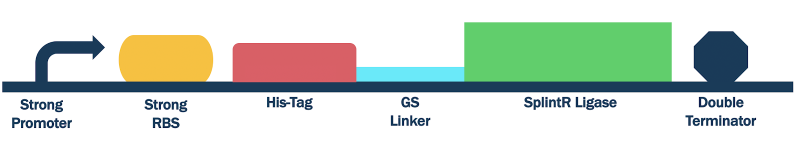
Figure #4. Design Strong Promoter SplintR Ligase Expressing Construct (BBa_K3352004).
Due to the usage of a promoter that isn’t T7, we used both DH5⍺ E. coli cells for both plasmid replication and protein expression (Arnaud-Barbe, 1998; Biolabs, n.d.; T7 Promoter System Vectors for Highest Expression Levels in Bacteria, n.d.). IPTG also served no purpose in inducing protein expression as T7 RNA Polymerase is not relied on (Biolabs, n.d.). Beside these alterations, the transformation, culturing, protein expression and purification were conducted in the same manner.
Characterization:
1. Construction of the T7 Promoter Library
A T7 promoter library was constructed through site-saturation mutagenesis, and an efficient screening method based on GFP fluorescence detection was established.
The site-saturation mutagenesis DNA fragment pET30a(+)-PT7-RFP vector 1 was obtained using the plasmid pET30a(+)-PT7-RFP as the template and vector 1.F (the sequence containing the mutation in promoter)and vector 1.R as the primers. At the same time, vector 2.F and vector 2.R as the primers to obtain pET30a(+)-PT7-RFP vector 1 by PCR. And then construct pET30a(+)-PT7 mutagenesis -RFP with pET30a(+)-PT7-RFP vector 1 and vector 2 by Gibson Assembly.

Fig 2. Site-saturation mutagenesis was conducted to obtain the T7 mutants
vector 1.F |
CCCGCGAAATTAATACGACTCACTNNNNNGGAATTGTGAGCGGATAAC |
vector 1.R |
TACCGCACAGATGCGTAAG |
vector 2.F |
TTCTCCTTACGCATCTGT |
vector 2.R |
AGTGAGTCGTATTAATTTCG |
Table 1: Primers for site-saturation mutagenesis
2. T7 Mutant Screening and Determination of GFP Fluorescence
T7 mutants were preliminarily selected by fluorescence of colonies grown on LB agar plates with Lactose. These mutants were inoculated in 2ml LB medium and cultured at 37 °C with agitation at 250 rpm for 12 h. Then, 1:100 inoculate to 200μl LB medium (with 0.2mM IPTG) in 96 - well plates at 30°C with agitation at 250 rpm for 16 h. The excitation wavelength was set to 553 nm, and the emission wavelength was set to 583 nm. The optical density of bacteria at 600 nm was detected with a spectrophotometer.
Finally, we have screened 8 different strength T7 mutants. The following figure is fluorescence/OD600 of them.

Fig. 2: fluorescence/OD600 of T7 mutants
Reference:
[1] Nie, Z., Luo, H., Li, J. et al. Appl Biochem Biotechnol (2019). https://doi.org/10.1007/s12010-019-03113-y
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
//direction/forward
//chassis/prokaryote/ecoli
//promoter
//regulation/constitutive
//chassis/bacteriophage/T7
| negative_regulators | |
| positive_regulators |