
 1 Registry Star
1 Registry StarPart:BBa_E2050
derivative of mRFP1, yeast-optimized
mRFP derivative. Ex548nm/Em562. yeast codon optimized.
Usage and Biology
mOrange, a mRFP (DsRed) derivative. Higher quantum yield but blue shifted and slower maturing compared to current (1/05) best red, mCherry.
We used the part BBa_E2050to construct mRFP-fused protein. Sequencing result revealed this part is working.
Tianjin IGEM Team
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21INCOMPATIBLE WITH RFC[21]Illegal BglII site found at 763
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
The followings were edited by SCU_China-2017
Improvement
Based on the part BBa_E2050 which containing coding sequence of mOrange only, we added pTetR promotor(BBa_R0040), RBS(BBa_B0034) and double terminators(BBa_B0010 and BBa_B0012) to it.
Characterization
In order to observe the fluorescence of mOrange easily and obviously, we transformed plasmid containing this part into E. coli BL21(DE3) strain which doesn’t have tetR gene. In this circumstance, mOrange can be expressed in BL21(DE3) cells constitutively.
After transformation, BL21(DE3) cells were cultured in LB solid media containing chloramphenicol for about 18 hours. We used two controls in the whole process, BL21(DE3) competent cells transformed with recombinant plasmid pCI-luxI-pSB1C3 and BL21(DE3) competent cells transformed with nothing.
BL21(DE3) colonies transformed with part BBa_K2276009 are orange. BL21(DE3) colonies transformed with pCI-luxI-pSB1C3 are in normal color. BL21(DE3) cells without any other plasmids can’t grow on plate containing chloramphenicol(Figure 1, a).
And then, we isolated the single colonies from selective plates, and inoculated a culture of about 3 mL LB medium containing chloramphenicol. The cultures were incubated at 37℃ for about 24 hours. Culture of BL21(DE3) cells transformed with part BBa_K2276009 are orange obviously. And BL21(DE3) cells transformed with pCI-luxI-pSB1C3 are light yellow(Figure 1, b).

The following characterizations were edited by the iGEM-team Goettingen-2018

Previous work using BBA_E2050 has only focused on its function in yeast. The aim of the experiments was to transform mERP into different Bacillus strains. Because the plasmid pSB1C3 does not contain an origin of replication for Bacillus, we cloned the fluorophore into the plasmid pAC7 and transformed it into competent DH5α. The fluorophore was additionally coupled to a self-made promoter, which is characterized by a perfect consensus sequence and perfect RBS for Bacillus. The E. coli strain has an increased transformation efficiency, where plasmid insertion is enhanced due to mutations in recA1 and endA1. pAC7 is a vector for the construction of translational lacZ fusions that can be integrated at the amyE site. Transformation with pAC7 allows direct integration of the fluorophore into the genome of Bacillus. In a further step, mERP was transformed into WT Bacillus subtilis strain 168, BP233 (trpC2 gltT::spc) and BP235 (trpC2 gltT::spc gltP::cat), which are characterized by a resistance towards the herbicide glyphosate.
Successful integration was verified by determination of α-amyE activity. The integration of the promoter-reporter gene fusion results in the inactivation of the amyE gene and the lack of α-amylase activity. As the functionality of the amyE gene can easily be determined by monitoring the activity of α-amylase, the lack of this enzymatic activity is a good indication for the correct integration of the promoter-reporter gene fusion. Furthermore, cells were grown in LB medium over night and cells were collected by centrifugation at 5000 rpm for 10 minutes. Pellets show the distinct orange colouration of mERP (Figure 1.)

Successful integration was verified by determination of α-amyE activity. The integration of the promoter-reporter gene fusion results in the inactivation of the amyE gene and the lack of α-amylase activity. As the functionality of the amyE gene can easily be determined by monitoring the activity of α-amylase, the lack of this enzymatic activity is a good indication for the correct integration of the promoter-reporter gene fusion. Furthermore, cells were grown in LB medium over night and cells were collected by centrifugation at 5000 rpm for 10 minutes. Pellets show the distinct orange colouration of mERP (Figure 1.)
Activity of the fluorophore in the WT 168 was further analysed with fluorescence microscopy. Therefore, strains were incubated overnight at 37°C with agitation in darkness. 10 µL of the culture was transferred on microscope slides with solid 1% agarose in H2O to lock the cells in place for long exposure times (Figure 2).

The effects of the fluorophore on the fitness of the different strains were analysed using a platereader. Cell growth was measured at an absorbance of λ=600 nm. Fluorescence activity was measured at an excitation wavelength of λ=529 nm and an emission at λ=562 nm. The fitness was calculated relative to the fitness of the WT strain. The results show that the fluorophores do not impact the growth of the strains (Figure 3) in any way and that fluorophores work perfectly in the presence of the self-made promoter. The tagged strains were used for further experiments to create a rapid test for detection of glyphosate.
Characterization by St_Andrews_2019
Figure 3 shows a small-scale Ni purification of yeast-optimised mOrange. Bands at the expected mass 26.7kDa, as well as a visibly Orange purified solution (Figure 4) indicated correct expression of mOrange. However, purified bands at ~20kDa and 10kDa indicate cleavage at (at least) two sites in the protein (not one site as only the N-terminus has a His tag). As the purified mOrange+SpyTag solution was not visibly orange and at half the expected mass, we suggest that it too has been cleaved.
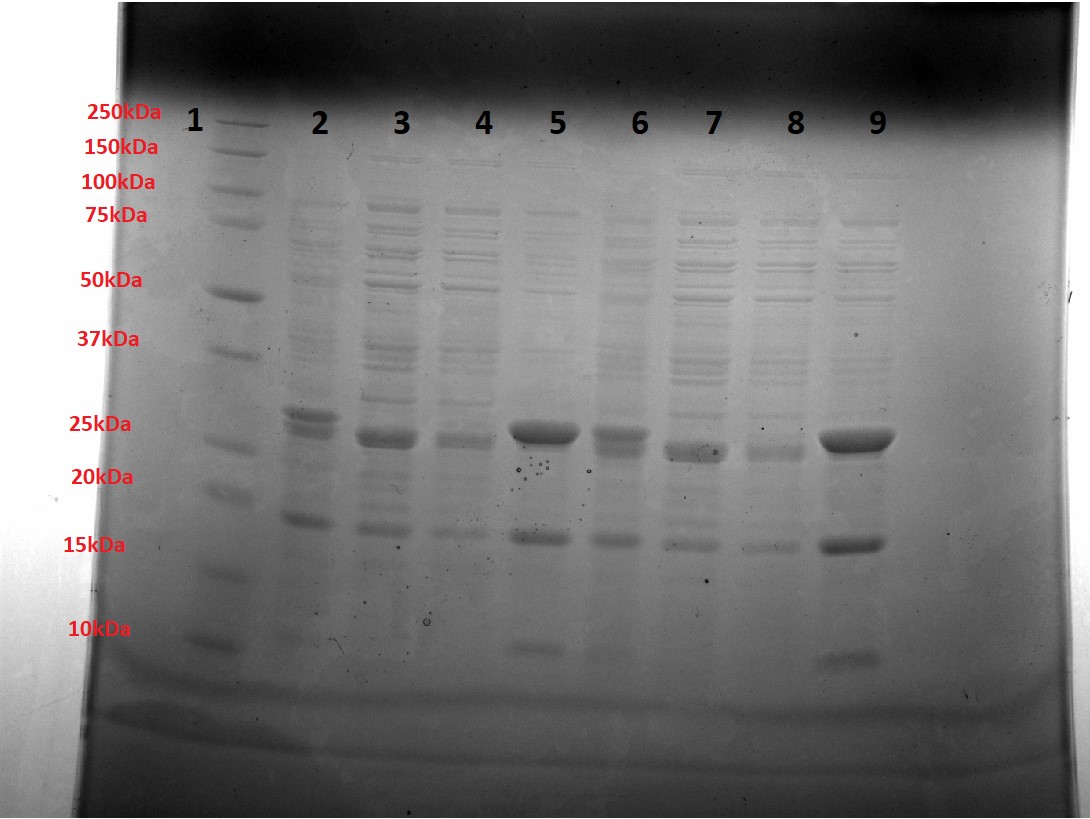
Figure 3: Small-scale Ni purification of mOrange. Lane 1 = Ladder, Lanes 2,6 = Unpurified sample, Lanes 3,7 = Flow-through sample, Lane 4,8 = Wash sample (5mM Imidazole), Lane 5,9 = Elution sample (250mM Imidazole).

Figure 4: The elution samples for mOrange, visibly orange.
To check the orange colour comes from the correct absorbance, we took an absorbance spectrum of the elution sample (Figure 5). We see a small peak at 548nm, the correct excitation wavelength for mOrange. Combining the correct expression weight and absorbance, we conclude successful expression of mOrange.
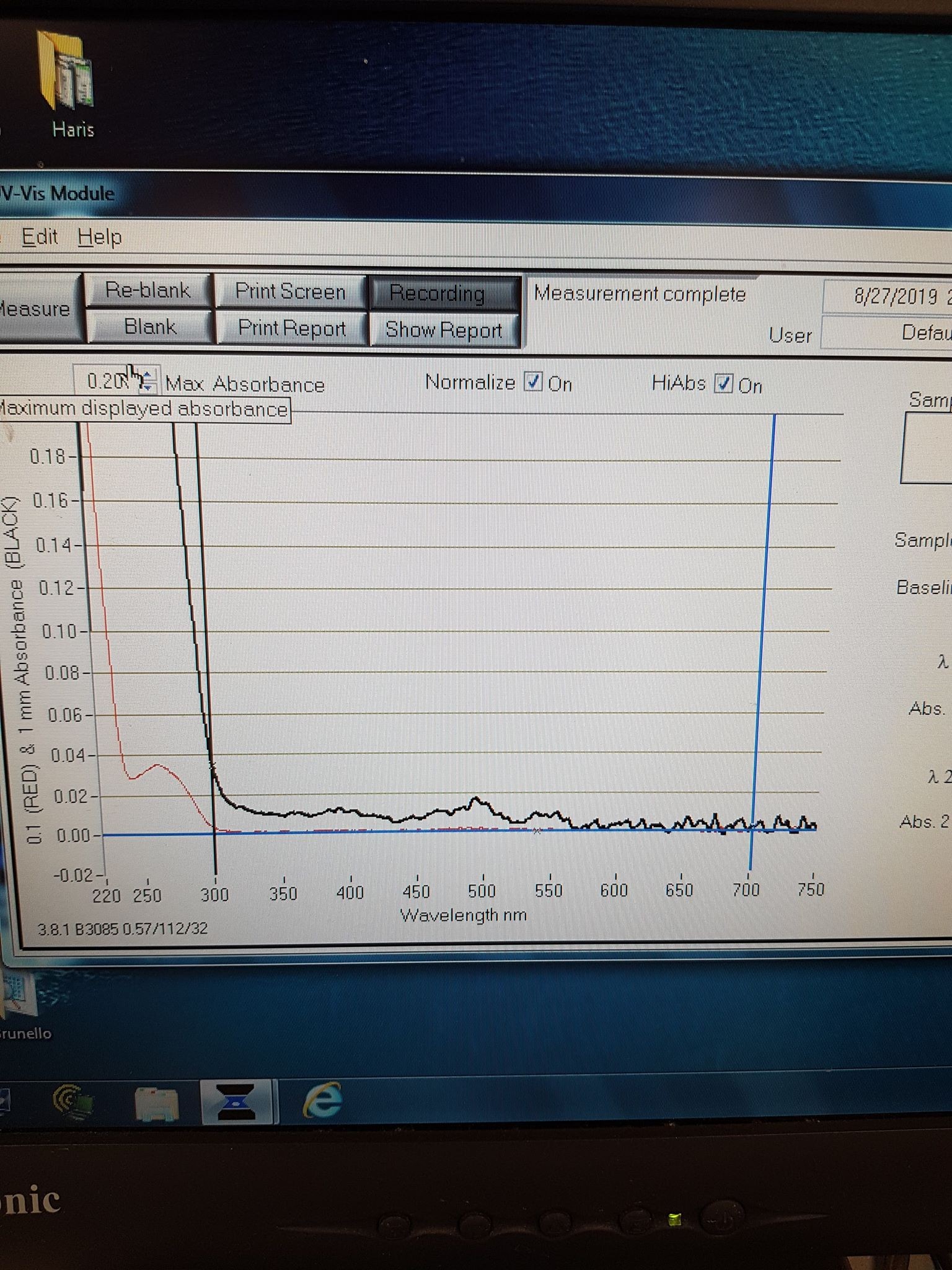
Figure 5: Absorbance spectrum for mOrange elution sample.
Estonia_TUIT 2021 team contribution
Monomeric orange fluorescent protein (mOrange)
mOrange is an extremely bright orange fluorescent protein that can be expressed both in bacterial and in mammalian cells (Kremers et al., 2009). It has a high extinction coefficient and quantum yield which allows it to be a perfect FRET (fluorescence resonance energy transfer) acceptor. mOrange has a slow half-life time of 2.5 hours for maturation at 37°C (mOrange :: Fluorescent Protein Database). Due to a low photostability (6.4 s) which is approximately 5% that of EGFP, its usage is limited. However, by applying a directed evolution approach that selected for higher photostability, this deficiency was overcome. This improvement resulted in the mOrange2 derivative, which has a photostability that is 30% higher than that of EGFP (Formation Stages of the mOrange Fluorescent Protein Chromophore).

Figure 1. The emission and excitation spectra of mOrange (mOrange:: Fluorescent Protein Database). It can be seen on Figure 4 that the excitation maximum for the mOrange is at 548 nm, and the emission maximum is at 562 nm. The extinction coefficient was 71000 M-1cm-1. Quantum yield is 0.69.
In the Nikon microscope settings for mOrange imaging were as in Table 1 (Imaging Fluorescent Proteins | Nikon’s MicroscopyU).
Table 1. Fluorescent Nikon microscope A1 HD25/A1R HD25 settings for mOrange (Imaging Fluorescent Proteins | Nikon’s MicroscopyU).
| Excitation Laser (nm) | Excitation Filter CWL / BW (nm) | Dichromatic Mirror Cut-On (nm) | Barrier Filter CWL / BW (nm) | Relative Brightness (% of EGFP) |
| He-Ne (543) | 525/40 | 550LP | 585/50 | 146 |
References
- Kremers, G. J., Hazelwood, K. L., Murphy, C. S., Davidson, M. W., & Piston, D. W. (2009). Photoconversion in orange and red fluorescent proteins. Nature Methods, 6(5), 355–358. https://doi.org/10.1038/NMETH.1319
- mOrange :: Fluorescent Protein Database. (n.d.). Retrieved October 16, 2021, from https://www.fpbase.org/protein/morange/
- Formation Stages of the mOrange Fluorescent Protein Chromophore. (n.d.). Retrieved October 16, 2021, from https://www.zeiss.com/microscopy/us/solutions/reference/all-tutorials/fluorescent-protein/morange-fluorescent-protein-chromophore-formation.html
- Imaging Fluorescent Proteins | Nikon’s MicroscopyU. (n.d.). Retrieved October 15, 2021, from https://www.microscopyu.com/techniques/fluorescence/fluorescent-protein-imaging-parameters
//chassis/eukaryote/yeast
//function/reporter/fluorescence
| abs | |
| color | Orange |
| emission | |
| emit | 562 |
| excitation | |
| excite | 548 |
| lum | |
| protein | mOrange |
| tag | None |