
Difference between revisions of "Part:BBa J04450"
YuchenZhou (Talk | contribs) |
|||
| (60 intermediate revisions by 10 users not shown) | |||
| Line 1: | Line 1: | ||
| − | + | ||
<partinfo>BBa_J04450 short</partinfo> | <partinfo>BBa_J04450 short</partinfo> | ||
| Line 109: | Line 109: | ||
Image:T--Tec-Chihuahua--RFP.jpeg | <i>E.Coli</i> SHuffle colonies transformed with BBa_J04450 | Image:T--Tec-Chihuahua--RFP.jpeg | <i>E.Coli</i> SHuffle colonies transformed with BBa_J04450 | ||
</gallery> | </gallery> | ||
| + | |||
| + | == Alma 2022 Improvement: Change to plasmid copy number, Double Inverter == | ||
| + | |||
| + | This year, we planned two improvements to the commonly used part, J04450. To better control the sometimes leaky expression, or to maximize expression, we created mutants of the origin sequence present on all pSB1C3 backbones. We introduced mutations +1, +8, -11, -13 from Camps et al, 2010, and the A715G reversion mutant from Lin-Chao et al, 1992 (turning this back into pBR322, instead of pUC19). | ||
| + | |||
| + | We collected plasmid copy number information using a qPCR protocol, and different approaches to gel quantitation (such as those found in Lin-Chao et al 1992 (and the variation present on the Barrick lab protocols page), Pushnova et al 2000. Here are some representative results: | ||
| + | |||
| + | [[File:T--Alma--wtpcn.png|650px|center]] | ||
| + | |||
| + | Based on several methods, we estimated the following ratio of plasmid to genomic DNA in a cell: | ||
| + | |||
| + | J04450: 76 | ||
| + | K4166002: 107 (Lin-Chao A715G) | ||
| + | K4166004: 307 (Camp +1) | ||
| + | K4166005: 381 (Camp +8) | ||
| + | K4166006: 133 (Camp +8 and -11) | ||
| + | K4166007: 153 (Camp -11) | ||
| + | K4166008: 194 (Camp +13) | ||
| + | |||
| + | In exponential cells, there is typically more than one copy of the genome present per cell, since the cells are actively replicating. Therefore, we would except to double or triple the above numbers to get a true plasmid copy number per cell. (This is based on the data at found from the BIONUMB3R5 database: https://bionumbers.hms.harvard.edu/bionumber.aspx?id=106438&ver=2&trm=Escherichia+Coli+Genome&org=) | ||
| + | |||
| + | We also tested the effect plasmid copy number had on expression of the RFP gene. Our math model takes this into account and suggested that at higher concentrations expression would become leaky. | ||
| + | |||
| + | [[File:T--Alma--pcnPlate.png|700px|center]] | ||
| + | |||
| + | From this, we can see that some of the mutations did increase or decrease expression. Some of the results were suprising - increased copy number abolished expression in some cases, presumably because DNA polymerase is replicating the plasmid so much that it cannot express the gene. | ||
| + | |||
| + | <b>Double Inverter (K4166010)</b> | ||
| + | |||
| + | We also planned an improvement by inserting a double inverter system between the lac promoter and the RFP gene. Our hope is that including this would prevent leaky expression by changing the dynamics of gene expression while preserving the logic of the BioBrick (turn on with IPTG). | ||
| + | |||
| + | We created two such parts: K4166009 and K4166010 - both have a Tet and cI inverter, but in different orders. | ||
| + | |||
| + | We tested their expression, both leaky and induced, in 0 and 100uM IPTG conditions: | ||
| + | |||
| + | [[File:T--Alma--dblinvplate.png|650px|center]] | ||
| + | |||
| + | As expected from our modeling, we found that our improved part has less leaky expression, although it has so far been difficult to induce expression. We may need to revisit some aspects of this design. | ||
== Alma 2020 Improvement: Successful Replication of reverted pSB1C3 in Vibrio Natriegens== | == Alma 2020 Improvement: Successful Replication of reverted pSB1C3 in Vibrio Natriegens== | ||
| Line 727: | Line 765: | ||
The results showed that after 16 hours, the number of cells was the largest and the medium became even deeper in red, indicating that 16 hours are at least required for productivity of the expressed protein in this conditions. It is quite likely that the promoter activity of the plasmid is most induced in the stationary phase. | The results showed that after 16 hours, the number of cells was the largest and the medium became even deeper in red, indicating that 16 hours are at least required for productivity of the expressed protein in this conditions. It is quite likely that the promoter activity of the plasmid is most induced in the stationary phase. | ||
| − | |||
| − | [[image:T--Gunma-- | + | [[image:T--Gunma--RFP_timecourse.png|800px|thumb|left]] |
| + | |||
| + | |||
| + | <html><br><br><br><br><br><br><br><br><br><br><br><br><br><br><br></html> | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | |||
| + | ==improvement: NAU-CHINA 2021== | ||
| + | |||
| + | <html><a href='https://parts.igem.org/Part:BBa_K3735666'>(more details see our part registry)</a></html> | ||
| + | |||
| + | Bacterial 5’ untranslated regions as an important part of mRNA (5’ UTR), it involves in a complex regulation of gene expression; however, the exact sequence features contributing to gene regulation are not yet fully understood. But many researches have proved that 5’ UTR plays a central role in gene expression and protein production. At the DNA level, it involves in transcript formation for the interplay between promoter and initially transcribed sequences (ITS). But, at mRNA level, it influences transcript stability and translation because of secondary structure formation and so on. Therefor 5’ UTR is has a crucial contribution to the maintenance of a fine balance between transcription, transcript stability and translation. In the work of Simone Balzer Le, Ingerid Onsager <i>et al</i>, they design a novel 5’UTR-Dual UTR, utilizing the transcriptional and translational characteristics of 5’UTRs in a single expression cassette, which will improve protein expression greatly. | ||
| + | |||
| + | <p>The Dual UTR contains two different UTRs which separated by a spacer region (Fig1). The 5’ UTR proximal to the promoter originates from the transcriptional screening(Tr-UTR) which affects transcript formation, while the second 5’ UTR is identified from the translational screening (Tn-UTR) which influences transcript stability and translation. And the spacer region provides enough space for physical separation of mutations affecting transcription and translation. By using the Dual UTR, we can improve the protein expression greatly.</p> | ||
| + | |||
| + | |||
| + | <html> | ||
| + | <img src="https://static.igem.org/mediawiki/parts/f/f6/T--NAU-CHINA--improvement1.png"width="700" height=""width="350" height=""/> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure.1 Dual UTR(BBa_K3735020) </b></p> | ||
| + | |||
| + | |||
| + | Our design: | ||
| + | After learning about the Dual UTR, we want to utilize it to improve an existing measurement part so that subsequent researchers can detect fluorescence more efficiently and accurately. Then we chose the BBa_J04450 which expresses mRFP under the induction of IPTG. We intend to add the Dual UTR between mRFP(BBa_E1010) and lac promoter(BBa_R0010) to improve its expression. Thereupon, we construct the following part(BBa_K3735666). | ||
| + | |||
| + | <html> | ||
| + | <img src="https://2021.igem.org/wiki/images/6/6e/T--NAU-CHINA--Improvement.fig.2.jpg"width="700" height=""width="350" height=""/> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig.2 Improved mRFP expression element(BBa_K3735666) </b></p> | ||
| + | |||
| + | |||
| + | Characterization: | ||
| + | We constructed the expression plasmid(BBa_K3735666) by using homologous recombination, which with Dual UTR sequence inserted. Then the recombinant plasmid was transformed into <i>E.coli</i> BL21(DE3) competent cells and grown overnight. Then the single colony(BBa_K3735666) and control(BBa_J04450) were used to inoculate in 50ml LB medium containing chloramphenicol and cultured to the same state. We cultured them under the induction of IPTG, and measured OD<sub>600</sub> and fluorescence intensity per 2 hours. | ||
| + | |||
| + | <html> | ||
| + | <img src="https://static.igem.org/mediawiki/parts/9/9f/T--NAU-CHINA-GENGGAI.png"width="700" height=""width="350" height=""/> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig.3 Plasmid map(A: Improved part (BBa_K3735666); B:Original part(BBa_J04450)) </b></p> | ||
| + | |||
| + | |||
| + | |||
| + | Results: | ||
| + | |||
| + | <html> | ||
| + | <img src="https://2021.igem.org/wiki/images/6/6b/T--NAU-CHINA--intu.png"width="700" height=""width="350" height=""/> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig.4 The results of improvement. A: the value of OD<sub>600</sub> of BBa_J04450 and BBa_K3735666; B: the fluorescence intensity of BBa_J04450 and BBa_K3735666; C: the fluorescence intensity/OD<sub>600</sub> of BBa_J04450 and BBa_K3735666 </b></p> | ||
| + | |||
| + | |||
| + | We can see that the fluorescence intensity of the improved part(BBa_K3735666) is significantly higher than that of the control group(BBa_J04450). Increasing the expression of fluorescence intensity can amplify weaker signals. At the same time, our method is also suitable for improving the expression intensity of other parts. | ||
| + | |||
| + | <html><a href='https://parts.igem.org/Part:BBa_K3735666'>(more details see our part registry)</a></html> | ||
| + | |||
| + | |||
| + | ==Improvement: NAU-CHINA 2022== | ||
| + | <partinfo>BBa_K4164020 short</partinfo> | ||
| + | |||
| + | The Ssr tag exists in bacteria extensively, serveing as a central feature of protein-quality control system. When protein translation stalls, the ribosome-rescue system mediates the addition of the sequence AANDENYALAA, an Ssr tag, to the C-terminus of the nascent polypeptide. The peptide sequence will lead the modified protein to degradation. In addition, Tobacco Etch Virus (TEV) protease is a site-specific protease which has the ability to recognize the amino acid sequence consisting of ENLYFQS and cleave it between Q and S. Taking advantage of the characteristics of TEV protease and Ssr tag, we can integrate a logic operation to decrease the leakiness of parts. | ||
| + | |||
| + | P<sub>lac</sub> promoter is a widely used promoter in induced expression of proteins. The gene products of <em>lacI</em> is a repressor for <em>lac</em> operon which can bind to <em>lac</em> operator, then block the RNA polymerase from binding to the P<sub>lac</sub> promoter and switching on transcription. When induced by IPTG, LacI can be removed from the site so that the transcription of downstream gene will be turned on. BBa_J04450 is a Red Florescence Protein (RFP) expression device driven by Plac promoter. When it works in <em>E.coli</em> , a quantity of RFP will express without IPTG due to the low expression level of LacI in the chassis. JNFLS-2020 has managed to reduce the leakiness by linking <em>lacI</em> and RFP to the downstream of P<sub>lac</sub>. This year, NAU-CHINA tried to build an anti-leakage device on BBa_J04450 which can be stretched to extensive inducible promoter. | ||
| + | |||
| + | |||
| + | |||
| + | The improvement of NAU-CHINA this year is to integrate TEV protease and Ssr tag to construct BBa_K4164020 in order to control the degradation of mRFP and resolve the leakage problem of BBa_J04450. | ||
| + | |||
| + | TEV cleavage site (ENLYFQS) and Ssr tag (AANDENYAAV) are inserted into the C-terminus of RFP in turn (Fig.1). Meanwhile, the TEV protease is expressed also driven by P<sub>lac</sub>. The Ssr tag can lead to the degradation of RFP expressed without IPTG. When induced by IPTG, the TEV protease can be inductively expressed, and cleave specifically at the cleavage site, which results in the loss of Ssr tag in RFP, and RFP lost the degradation tag will no longer be degraded and begin to work. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4164/wiki/part-registry/part-020-1.png"with="700" height="" width="350" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 1. Improvement of mRFP expression device.</b></p> | ||
| + | |||
| + | We used pET29a(+) as the vector, which is capable of expressing LacI to conduct BBa_K4164020. Additionnaly, we constructed BBa_J04450, BBa_K4164021 and BBa_K4164022 (Fig.2) in pET29a(+) as control groups. These plasmids were transformed into <em>E.coli</em> BL21 respectively and grown in LB medium at 37℃ until the optical density at 600nm (OD600) reached 0.6. After that, the bacteria culture fluid was incubated in 50mL LB medium with or without IPTG induction at 37℃ for 12 hours, and measured the OD600 and fluorescence intensity (excitation 535 nm and emission 605 nm) per hour. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4164/wiki/improvement/improvement-fig-2-png.png"with="1000" height="" width="700" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 2. a.Improved part BBa_K4164020; b. Original part BBa_J04450; c. Control part 1 BBa_K4164021; d. Control part 2 BBa_K4164022</b></p> | ||
| + | |||
| + | |||
| + | |||
| + | We find that the leakage level of BBa_K4164020 is lower compared with BBa_J04450 and BBa_K4164021. What’s more, due to the absence of TEV protease, the fluorescence intensity of BBa_K4164022 is poor regardless of whether there is IPTG induction. However, the relative fluorescence intensity of BBa_K4164020 and BBa_J04450 are similar in the condition of IPTG induction, which suggests the Ssr tag has little influence on the expression of RFP under normal circumstances. | ||
| + | |||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4164/wiki/improvement/improvement-fig-3-png.png"with="1000" height="" width="700" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"> | ||
| + | <b>Figure 3. the result of improvement. <br> | ||
| + | a. the variation of fluorescence intensity/OD of BBa_K4164020, BBa_J04450, BBa_K4164021 and BBa_K4164022 cultivated with or without IPTG induction;<br> | ||
| + | b. the fluorescence intensity/OD of BBa_K4164020, BBa_J04450 and BBa_K4164021 cultivated without IPTG induction;<br> | ||
| + | c. the fluorescence intensity of BBa_K4164020, BBa_J04450, BBa_K4164021, BBa_K4164022 and Control cultivated with IPTG after 12 hours;<br> | ||
| + | d. the fluorescence intensity of BBa_K4164020, BBa_J04450, BBa_K4164021, BBa_K4164022 and Control cultivated without IPTG after 12 hours.<br></b></p> | ||
| + | |||
| + | ==Contribution: NYCU_Formosa 2022== | ||
| + | ===Color mixing model=== | ||
| + | To establish our color mixing model, we measured the fluorescent intensity to time relationship of Part: BBa_J04450(rfp) and Part: BBa_K741002(gfp). With these parameters, we created a model for future teams to deduce the ratio of red and green for their desired color. This simulation shows that different RGB values lead to different color results. What’s more important is that the changing degree of red and green values is based on our protein expression experiments. Thus, according to their desired color, future teams are able to speculate the specific incubation time for ''E.coli'' transformed with RFP and GFP genes. | ||
| + | |||
| + | With experimental verification, we have mixed different concentrations of RFP and GFP in fixed value of 200μL, which includes ratio 1:1, 5:3, 2:1, 3:1, 4:1, 9:1, 1:9, 4:1, 3:1, 2:1. | ||
| + | |||
| + | [[File:Nycu Formosa GFP.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 1. RFP : Flourscecnce Intensity of RFP over time</b></p> | ||
| + | |||
| + | [[File:Nycu Formosa RFP.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 2. GFP : Flourscecnce Intensity of GFP over time</b></p> | ||
| + | |||
| + | [[File:NYCU Formosa Fig1.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 3. Different combinations of RFP and GFP</b></p> | ||
| + | |||
| + | [[File:NYCU Formosa fig2.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 4. Combinations of RFP and GFP of 1:9 and 9:1.</b></p> | ||
| + | |||
| + | [[File:Nycu formosa fig3.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 5. Combinations of RFP and GFP of 1:5 and 5:1.</b></p> | ||
| + | |||
| + | [[File:Nycu formosa fig4.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 6. Combinations of RFP and GFP of 1:4 and 4:1.</b></p> | ||
| + | |||
| + | [[File:Nycu formosa fig5.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 7. Combinations of RFP and GFP of 1:3 and 3:1.</b></p> | ||
| + | |||
| + | [[File:Nycu formosa fig6.jpg | center]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 8. Combinations of RFP and GFP of 1:2 and 2:1.</b></p> | ||
| + | |||
| + | [[File:Nycuformosa1111.png | center | 700px]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 9. Color Mixing Model 1.</b></p> | ||
| + | |||
| + | [[File:Nycuformosa3333.png | center | 700px]] | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Figure 10. Color Mixing Model 2.</b></p> | ||
| + | |||
| + | |||
| + | ==Team NAU-CHINA 2023 contribution: Effect of mRFP expression on the cell abundance measurements== | ||
| + | |||
| + | ====Purpose & Design==== | ||
| + | In our contribution section, we decided to delve deeper into the investigation of the commonly used reporter gene, Red Fluorescent Protein (RFP). Red Fluorescent Proteins (RFPs) possess a strong absorption of light at the 600 nm wavelength, leading to a misrepresentation of cell optical density and consequently an underestimation of single-cell fluorescence. Thus, we selected BBa_J04450 from the iGEM part repository and used pSB1C3 as the vector to introduce it into <i>Escherichia coli</i> DH5α for expression, aiming to explore the influence of different light wavelengths on the deviation in measuring Red Fluorescent Protein (RFP) abundance in cells. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4613/wiki/parts/plasmid-map-of-psb1c3-j04450.png"with="700" height="" width="700" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig. 1 The Plasmid Map of pSB1C3-J04450. | ||
| + | </b></p> | ||
| + | |||
| + | ====Experiment==== | ||
| + | 1. We activated BBa_J04450 obtained from the kit and transformed it into <i>E. coli</i> DH5α, followed by overnight incubation at 37℃. The subsequent day, we chose 4 colonies and cultured them in 5 mL LB medium for approximately 12 hours. This genetic segment was expressed within the plasmid pSB1C3; consequently, both our LB agar plates and culture media were supplemented with chloramphenicol (50 μg/mL). | ||
| + | |||
| + | 2. Used XbaⅠ and SpeⅠ enzymes to construct the empty vector pSB1C3. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4613/wiki/parts/psb1c3-plasmid.png"with="700" height="" width="700" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig. 1 The Plasmid Map of pSB1C3. | ||
| + | </b></p> | ||
| + | |||
| + | 3. The pSB1C3 plasmid was transformed into <i>E. coli</i> DH5α, followed by overnight incubation at 37℃. The next day, four colonies were selected and cultured in 5 mL LB medium for approximately 12 hours. | ||
| + | |||
| + | 4. We prepared six fresh 50 mL LB media. Subsequently, we introduced overnight cultures of <i>E. coli</i> carrying pSB1C3-J04450 and pSB1C3 , respectively, into these media. The initial three media were inoculated with pSB1C3 <i>E. coli</i>, while the latter were pSB1C3-J04450 <i>E. coli</i>. These cultures were cultivated for sixteen hours. | ||
| + | During the sampling process, we transferred 200 μL cultured samples into Costar 96 Flat Bottom Polystyrene Cat with black and transparent bottoms. Notably, each sample was subjected to three replicates within both the black and transparent groups. | ||
| + | |||
| + | 5. After sampling, we promptly measured the data using an microplate reader (Tecan Spark) and conducted subsequent analysis. | ||
| + | |||
| + | ====Results==== | ||
| + | Among commonly used fluorescent proteins, the excitation wavelength for mRFP is situated around 585 nm (Fig. 2). Consequently, it exhibits a prominent absorption peak at 600 nm (Fig. 2), a standard wavelength used for measuring bacterial culture optical density (OD). This phenomenon leads to a significant overestimation of data when quantifying the abundance of bacteria expressing red fluorescent protein. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4613/wiki/parts/parts/absorption-spectra-of-mrfp1.jpeg"with="1000" height="" width="500" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig. 2 Absorption spectra of mRFP1. | ||
| + | </b></p> | ||
| + | |||
| + | Initially, we measured the fluorescence levels of <i>E. coli</i>. Notably, the <i>E. coli</i> carrying pSB1C3-J04450 exhibited apronounced shift towards red fluorescence in the later stages. In the early stages, <i>E. coli</i> carrying pSB1C3-J04450 displayed fluorescence levels similar to those without it. However, after 12 hours, a substantial increase was observed, significantly surpassing the fluorescence levels of the <i>E. coli</i> carrying pSB1C3 (Fig. 3). This indicates the density of our experimental strain, which exhibits a distinct disparity compared to <i>E. coli</i> carrying pSB1C3. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4613/wiki/parts/parts/the-fluorescence-of-each-culture.png"with="1000" height="" width="750" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig. 3 The Fluorescence of each culture. | ||
| + | </b></p> | ||
| + | |||
| + | We measured the OD values of the bacteria, revealing distinct differences between <i>Escherichia coli</i> strains carrying pSB1C3-J04450 and those carrying pSB1C3 at various wavelengths. Specifically, data obtained under 585 nm wavelength exhibited the most significant disparities between the two <i>E. coli</i> strains, while data acquired under 700 nm wavelength displayed less obvious distinctions (Fig. 4a-d). The increased disparity at 585 nm can be attributed to the absorption peak of mRFP occurring near this wavelength, thus making the data differences between the two strains more pronounced. Thus, the influence of RFP can be disregarded at the two longer wavelengths. | ||
| + | |||
| + | Furthermore, to gain further insight into the impact of RFP on <i>E. coli</i> OD values, we conducted deviation analyses relative to the baseline at 700 nm and performed linear function fitting for the data obtained at the other three wavelengths. It is evident that the data acquired at 660 nm exhibited minimal deviation, while the other two shorter wavelengths displayed a noticeable upward trend (Fig. 4E). At the 12-hour mark, the OD values obtained at 600 nm were nearly 15% higher than those unaffected by RFP. This illustrates the impact of mRFP expression on the measurement of bacterial density under wavelength at 600 nm. This serves as a valuable reference for future teams. | ||
| + | |||
| + | <html> | ||
| + | <center><img src="https://static.igem.wiki/teams/4613/wiki/parts/parts/the-od-subabssub-and-deviation-analyses.png"with="1000" height="" width="750" height=""/></center> | ||
| + | </html> | ||
| + | |||
| + | <p style="text-align: center!important;"><b>Fig. 4 The OD<sub>abs</sub> and Deviation Analyses a-d OD<sub>abs</sub> measurements were taken at wavelengths of 585 nm, 600 nm, 660 nm, and 700 nm. Due to the absorption peak of RFP at 585 nm, <i>E. coli</i> carrying pSB1C3-J04450 exhibited significantly higher data values at both 585 nm and 600 nm wavelengths compared to those carrying pSB1C3. However, data at 660 nm and 700 nm showed relatively minor differences. e: Using 700 nm as reference, bias analysis and function fitting were conducted for the data obtained at the other three wavelengths. In comparison to OD at 660 nm, the OD at 585 nm and 600 nm displayed a notable positive correlation, highlighting the significant impact of RFP expression on bacterial concentration measurements at 585 nm and 600 nm. | ||
| + | </b></p> | ||
Latest revision as of 14:09, 11 October 2023
RFP Coding Device
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25INCOMPATIBLE WITH RFC[25]Illegal AgeI site found at 781
Illegal AgeI site found at 893 - 1000COMPATIBLE WITH RFC[1000]
The colonies are clearly red in color under natural light after about 18 hours. Smaller colonies are visibly red under UV. The RFP part does not contain a degradation tag and the RBS is strong.
- LacI sensitive
- CAP sensitive
Contents
- 1 Improvement by JNFLS2020
- 1.1 Overview:
- 1.2 Results:
- 1.3 Protocol:
- 1.4 Pictures
- 1.5 Alma 2022 Improvement: Change to plasmid copy number, Double Inverter
- 1.6 Alma 2020 Improvement: Successful Replication of reverted pSB1C3 in Vibrio Natriegens
- 1.7 Team Amazonas_Brazil 2019:The RFP sinalization system in E. coli Nissle 1917
- 1.8 Team NAU_CHINA 2019:The inhibitory effect of different concentrations of glucose in LB culture
- 1.9 Team ITB_Indonesia 2017: Red color dynamics of cloned Escherichia coli strains in LB broth
- 1.10 Team INSA-UPS France 2017 : usage in Vibrio harveyi strain engineered by conjugation
- 1.11 IIT Madras 2016's Characterization
- 1.12 Experimentation
- 1.13 Usage in Chromobacterium Violaceum
- 1.14 H14Z1_Hangzhous 2018's Characterization
- 1.15 SCAU-China 2018's improvement
- 1.16 Grenoble-Alpes 2019 Characterization
- 1.17 Tec-Chihuahua 2019 Characterization: RFP expression in E.Coli SHuffle strain
- 1.18 Team Lund 2019 charecterization
- 1.19 Team BUCT_China 2019
- 1.20 Newcastle 2019 characterisation
- 1.21 Team Aalto-Helsinki 2019 characterization: Effect of antibiotic concentration on fluorescence intensity
- 1.22 Team Botchan_Lab_tokyo 2019 characterization
- 1.23 description
- 1.24 Experimental Design
- 1.25 Result
- 1.26 conclusion
- 1.27 Contribution: Gunma 2021
- 1.28 improvement: NAU-CHINA 2021
- 1.29 Improvement: NAU-CHINA 2022
- 1.30 Contribution: NYCU_Formosa 2022
- 1.31 Team NAU-CHINA 2023 contribution: Effect of mRFP expression on the cell abundance measurements
Improvement by JNFLS2020
Overview:
BBa_J04450 is a RFP generator driven by Plac promoter, which is regulated by LacI and induced by IPGT. Plac is one of the most common promoter in life science research field. It is mainly composed of Lac operon containing LacO site. LacI repressor, encoded by LacI gene, can bind to LacO site, inhibiting the binding of RNA polymerase to the promoter. So the expression of downstream genes is blocked. Serving as inducer, IPTG can bind with LacI repressor, making the latter’s conformation changes, so LacI is detached from LacO site, which enables the transcription of downstream genes. BBa_J04450 is a RFP generator driven by Plac promoter, however there is no LacI gene in it. Although the E. coli could express some LacI, it is not enough for inhibition RFP expression. So this RFP generator has some leakage expression, like the designer stated that some RFP express even without IPTG inducer presence.
This year, we constructed a new RFP generator (BBa_K3605010) also driven by Plac promoter. It contains LacI gene, which can lower significantly the RFP leakage expression. Both LacI and RFP are linked to the downstream of Plac, which is regulated by both LacI repressor and IPTG inducer. Using RFP as a reporter, its fluorescence intensity appears a lower leakage expression. This new RFP generator could be self-regulated because LacI protein can inhibit its self-expression, so that no excessive LacI expression which is considered as waste of resources. We detected the response of this generator to different concentration of IPTG, indicating that it could be inhibited by LacI, and induced well by IPTG.
Results:
Overlapping PCR was used to clone the LacI and RFP genes. Then the PCR product was inserted to plasmid pSB1C3 to create the new part BBa_K3605010. The identification result is showed in Fig.1.

Fig.1. The result of BBa_K3605010 construction.
M: Marker; 1: Digestion of BBa_K3605010; 2: Plasmid of BBa_K3605010.
We compared the inducing effect of IPTG on the two RFP generators, using different concentration of IPTG. We set 5 groups: 4 experimental groups of the old RFP generator (BBa_Jo4450) and the new RFP generator (BBa_K3605010) with or without IPTG, one negative control without RFP expression. At 0h, all groups’ OD600 approximately reaches to 0.6, then certain concentration of IPTG was added to the culture medium, incubated cells at 37℃ for 20h. Measure the fluorometric value at 610 nm and OD600 value for each group every 2h, using an automatic microplate reader. The results are showed as follows (Fig.2-Fig.4).

Fig.2. The inducing effect of IPTG on the two RFP generators. Relative fluorescent intensity is fluorescence per OD600 standardized with fluorescence per OD600 value of each test group at time = 0, IPTG=0. The figure indicated that both BBa_J04450 and BBa_K3605010 expressed RFP induced by IPTG, and different concentration of IPTG had same inducing trend.
Then we detected the RFP expression with IPTG presence or absence, using BBa_J04450 and BBa_K3605010, respectively. The results showed that BBa_J04450 expressed a mountain of RFP whether IPTG is present or not, which means that BBa_J04450 had a high leakage expression and lower sensitivity to the IPTG induction. However, BBa_K3605010 expressed a high level RFP only with IPTG presence. The leakage expression is very low, and it is sensitive to the IPTG induction.

Fig.3. The comparison of RFP expression using BBa_J04450 with and without IPTG induction. Relative fluorescent intensity is fluorescence per OD600 standardized with fluorescence per OD600 value of each test group at time = 0, IPTG=0. This figure indicated that the RFP generator (BBa_J04450) had a high leakage expression, and low sensitivity to the IPTG induction.

Fig.4. The comparison of RFP expression using BBa_K3605010 with and without IPTG induction. Relative fluorescent intensity is fluorescence per OD600 standardized with fluorescence per OD600 value of each test group at time = 0, IPTG=0. This figure indicated that the new RFP generator (BBa_K3605010) is very sensitive to the IPTG induction, and the RFP leakage expression is very low.
Protocol:
- Transform the plasmids into BL21 strain.
- Pick up a single colony by a sterile tip from each of the plates for all the experimental and control groups. And put the colony into 5ml M9 medium with 100 µg/ml ampicillin. Incubate at 37℃ in a shaker for 20h.
- Detect OD600value of the culture medium with spectrophotometer, and dilute the culture medium to OD600= 0.6.
- Add 100 µl bacteria culture medium into a sterile 96-well plate. IPTG is added to final concentrations of 1, 2, 5, 10mM. M9 medium is the blank control. The colony without RFP expression is the negative control.
- Incubate at 37℃, and measure the fluorometric value at 610 nm and OD600value for each well every 2h, using an automatic microplate reader.
- The experiment should be repeated at least 3 times.
This part is commonly used, but can fail if the system contains LacI or CAP protein.
(--Meagan 15:39, 23 July 2009 (UTC))
[http://2012.igem.org/Team:TU_Munich Team TU_Munich 2012] improved this part by making it compatible to RFC10 and RFC25 (see: BBa_K801100)
(--VolkerMorath 15:02, 21 October 2012 (UTC))
[http://2013.igem.org/Team:NRP-UEA-Norwich Team NRP-UEA 2013] improved this part by adding a NdeI restriction site before the RFP gene. (see: BBa_K1041000)
(--holusac 20:46, 14 August 2013 (UTC))
[http://2015.igem.org/Team:Warwick Team Warwick 2015] improved this part by analysing the effect of copy number on gene expression.
(--Lcarroll 20:48, 25 September 2015 (UTC))
[http://2016.igem.org/Team:Leiden Team Leiden 2016] contributed to the characterisation of this part by showing equal functionality in simulated microgravity (0g) as in the normal gravity of the Earth.
(--Valentijn 19:38, 19 October 2016 (UTC))
[http://2017.igem.org/Team:UChicago Team UChicago 2017] contributed to this part by improving/changing the documented sequence through mutagenesis to create blunt-end restriction sites for cloning not within the prefix/suffix region (created BBa_K2428000).
(--pzulueta97 21:14, 25 October 2017 (UTC) )
[http://2017.igem.org/Team:Grenoble-Alpes Team Grenoble-Alpes 2017] contributed to the characterisation of this part by testing the time of apparition of fluorescence, in presence of IPTG or not (because the promoter leaks), as well as they contributed to the improvement of this part by using its fluorescence as a detection signal to be able to detect Vibrio Cholerae.
(--NoreenLouis 20:47, 26 October 2017 (UTC) )
[http://2017.igem.org/Team:Kingsborough_NY Team Kingsborough NY 2017] contributed to the characterization of this part by showing decreased fluorescence when expressed either in a higher salt media - such as LB with 3% sodium chloride - or E. coli that lacks tmRNA, the principal component of the cell's ribosome rescue system. View the data on the experience page or [http://2017.igem.org/Team:Kingsborough_NY/RFP visit our Wiki]
(--djcamenares 17:56, 27 October 2017 (UTC) )
[http://2017.igem.org/Team:iTesla-SoundBio Team iTesla SoundBio 2017] contributed to the characterization of this part by analyzing the rate of false positives when using the coloring of transformed colonies as a red/white screen for determining experimental success.
(--gladish 01:26, 28 October 2017 (UTC) )
[http://2018.igem.org/Team:Grenoble-Alpes Team Grenoble-Alpes 2018] contributed to the characterisation of this part by testing the delay before apparition of fluorescence directly after transformation and the intensity of the leak, in three different E. Coli strains.
(--perrine 15:06, 9 October 2018 (UTC) )
[http://2018.igem.org/Team:H14Z1_Hangzhou Team H14Z1_Hangzhou 2018] contributed to the characterisation of this part by testing the fluorescence idensity in different condition(e.g.temperature,medium volume, IPTG concentration) ,in two different E. Coli strains.
(--ericxu 09:32, 17 October 2018 (UTC) )
[http://2018.igem.org/Team:SKLMT-China Team SKLMT-China 2018] improved this part by replacing the induced promoter lacI with the strong constitutive pf-5 promoter PampC.(see: BBa_K2569029)
(--DDY 16:47, 17 October 2018 (UTC) )
Team Grenoble-Alpes 2019 contributed to the characterisation of this part by testing the effect of cAMP on the promoter’s leakages and protein expression in Escherichia coli.
(--pinerol 19:50, 27 September 2019 (UTC) )
Team Lund 2019 contributed to the charazterization of this part by testing the effect of different media composition in different E. coli strains on RFP expression.
(-- ParselTMatts 18:40, 19 October 2019 (UTC) )
Team BUCT-China 2019 Contribute to the characterization of this part by introducing it in Trans10 to observe its growth and draw its growth curve.
(-- buct_china_ljh 12:18, 20 October 2019 (UTC) )
Team Aalto-Helsinki 2019 contributed to the charazterization of this part by testing the effects of different chloramphenicol concentrations on RFP expression levels.
(-- Toivanen 15:20, 21. October 2019 (UTC) )
Team NAU_CHINA 2019 contributed to the characterization of this part by testing the effect of glucose on the protein expression in E. coli.
(-- NiuDan 17:41, 21. October 2019 (UTC) )
Team Botchan_Lab_Tokyo 2019 contributed to characterization of this part by testing radiation tolerance of E.coli express the RFP.
(-- youma 21:20, 21. October 2019 (UTC) )
Pictures
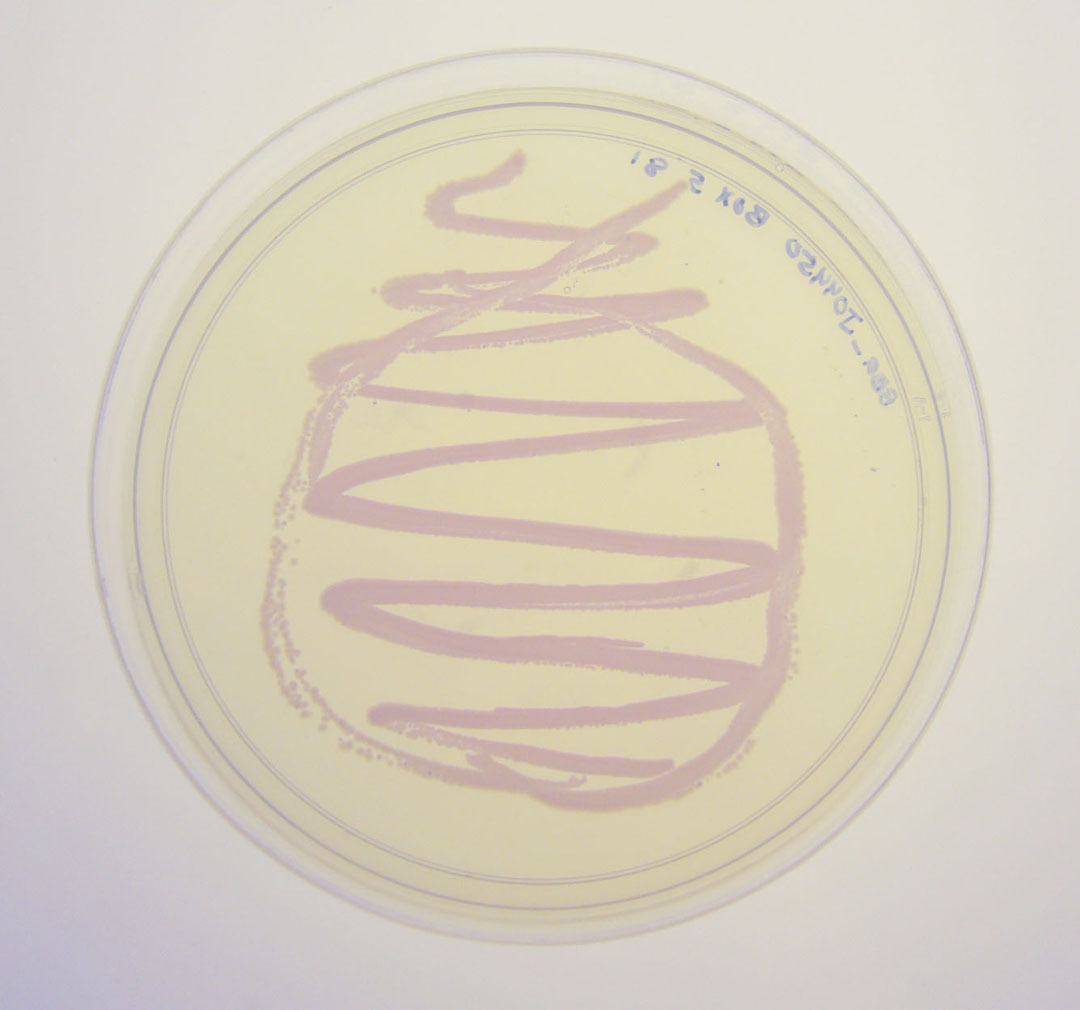
BBa_J04450 visualized under non-UV lightbox

BBa_J04450 visualized under 254nm wavelength UV lightbox
BBa_J04450 Colonies
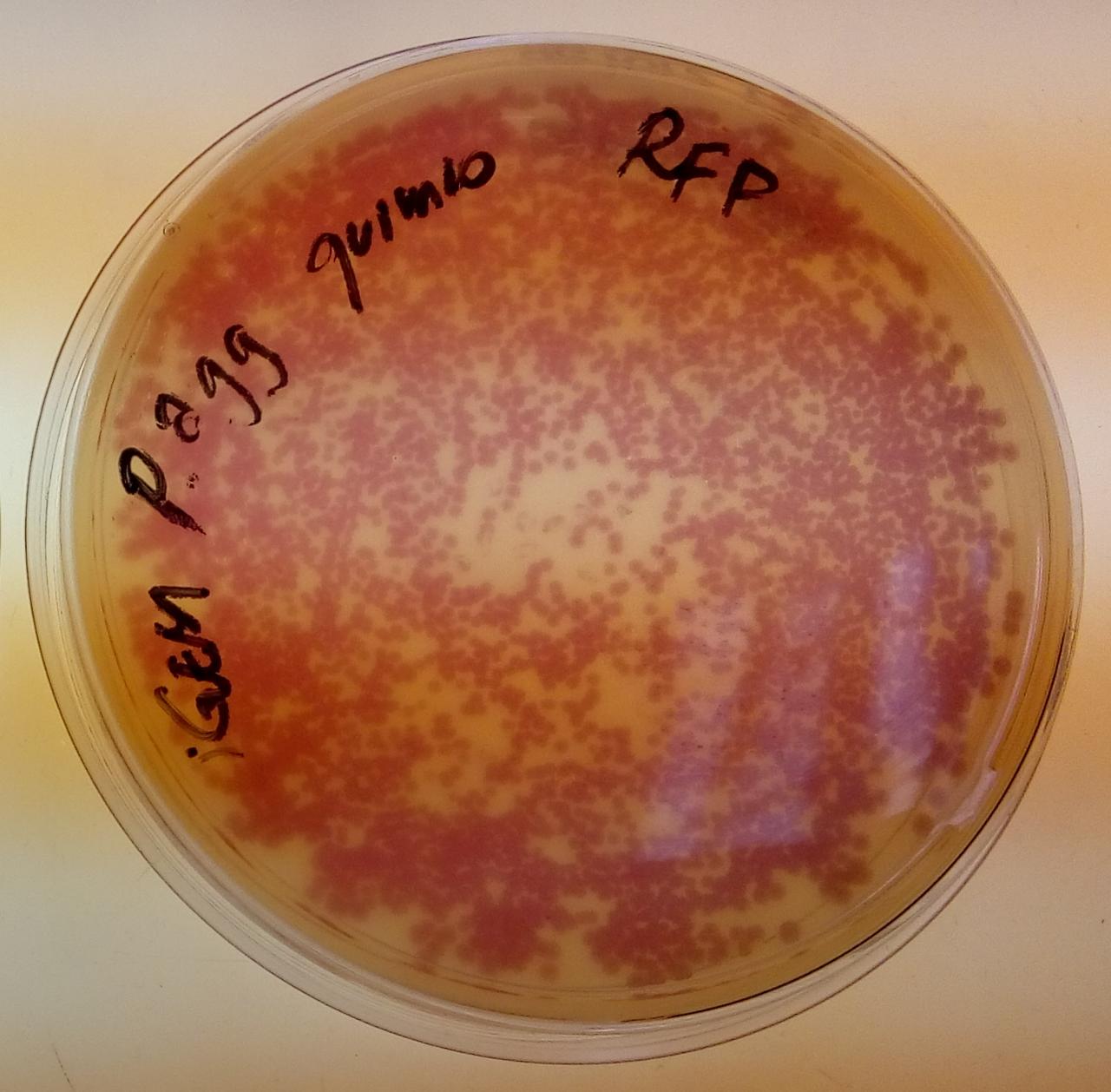
Pantoea agglomerans colonies in chloramphenicol, containing BBa_J04450 in the pSB1C3 backbone
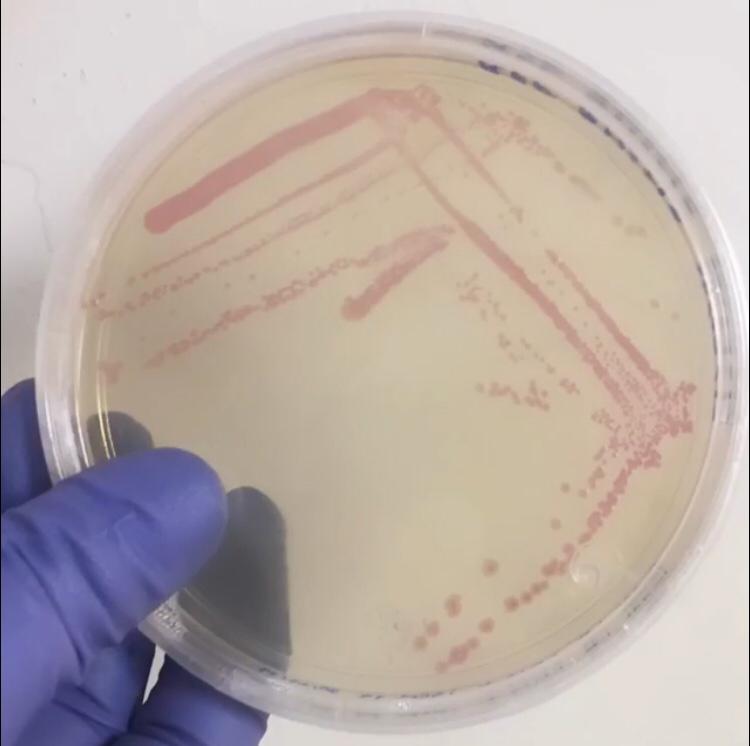
E.Coli SHuffle colonies transformed with BBa_J04450




Alma 2022 Improvement: Change to plasmid copy number, Double Inverter
This year, we planned two improvements to the commonly used part, J04450. To better control the sometimes leaky expression, or to maximize expression, we created mutants of the origin sequence present on all pSB1C3 backbones. We introduced mutations +1, +8, -11, -13 from Camps et al, 2010, and the A715G reversion mutant from Lin-Chao et al, 1992 (turning this back into pBR322, instead of pUC19).
We collected plasmid copy number information using a qPCR protocol, and different approaches to gel quantitation (such as those found in Lin-Chao et al 1992 (and the variation present on the Barrick lab protocols page), Pushnova et al 2000. Here are some representative results:

Based on several methods, we estimated the following ratio of plasmid to genomic DNA in a cell:
J04450: 76 K4166002: 107 (Lin-Chao A715G) K4166004: 307 (Camp +1) K4166005: 381 (Camp +8) K4166006: 133 (Camp +8 and -11) K4166007: 153 (Camp -11) K4166008: 194 (Camp +13)
In exponential cells, there is typically more than one copy of the genome present per cell, since the cells are actively replicating. Therefore, we would except to double or triple the above numbers to get a true plasmid copy number per cell. (This is based on the data at found from the BIONUMB3R5 database: https://bionumbers.hms.harvard.edu/bionumber.aspx?id=106438&ver=2&trm=Escherichia+Coli+Genome&org=)
We also tested the effect plasmid copy number had on expression of the RFP gene. Our math model takes this into account and suggested that at higher concentrations expression would become leaky.

From this, we can see that some of the mutations did increase or decrease expression. Some of the results were suprising - increased copy number abolished expression in some cases, presumably because DNA polymerase is replicating the plasmid so much that it cannot express the gene.
Double Inverter (K4166010)
We also planned an improvement by inserting a double inverter system between the lac promoter and the RFP gene. Our hope is that including this would prevent leaky expression by changing the dynamics of gene expression while preserving the logic of the BioBrick (turn on with IPTG).
We created two such parts: K4166009 and K4166010 - both have a Tet and cI inverter, but in different orders.
We tested their expression, both leaky and induced, in 0 and 100uM IPTG conditions:

As expected from our modeling, we found that our improved part has less leaky expression, although it has so far been difficult to induce expression. We may need to revisit some aspects of this design.
Alma 2020 Improvement: Successful Replication of reverted pSB1C3 in Vibrio Natriegens
We have been unable to transform J04450 into Vibrio natriegens, despite being able to get other ColE1 and pUC19 origin based plasmids in this species. We have discovered that pSB1C3 contains a point mutation in the origin of replication that could explain this effect. This part is a reversion of this mutation, making pSB1C3 into a true pUC19 origin plasmid. We proceeded to test this by transforming by electroporation into Vibrio natriegens, as well as measuring plasmid copy number.



The results above are representative of several experiments. As can be seen, J04450 cannot be transformed into Vibrio, while our mutant was able to successfully produce colonies. In these results, “-“ is the negative control plate streaked with pSB1C3 containing J04450 (“RFP”). This is the most commonly used backbone for iGEM BioBricks. The plate contains no growth. “+” contains pGGA without the plasmid that expresses RFP. This contains the pUC19 origin, which has expression in vibrio. However, pGGA is not compatible with iGEM BioBricks. “Mr” (BBa_K3445002) is the mutated backbone (BBa_K3445000) to both contain the gene needed to express RFP and allow for compatibility to show expression in vibrio. We observed in these experiments a red color in Vibrio, indicating that the bacteria can express the RFP gene.
We also measured the plasmid copy number of our mutant, both in E. coli and in Vibrio. We did so by performing qPCR on DNA isolated from equal numbers of these bacteria using either genome targeting primers (dnaE) or plasmid targeting primers (oriQ), as done in previous studies. The copy number is thus measured as 2 raised to the difference between the oriQ Ct value and the dnaE Ct value.



In E. Coli, the copies of plasmid per genome for J04450 with a pSB1C3 backbone was 6.92 copies as measured by qPCR. For the mutated backbone containing J04450 (“K3445002”), the plasmid per genome measured was 12.21, 25.11, and 10.85 copies. This averages to roughly 16.05 copies of plasmid per genome shown in e. Coli for K3445002. In vibrio, the reported values for plasmid per genome for pGGA was found to be 306 copies. The found values for copies of plasmid per genome for K3445002 in vibrio could not be determined in practice due to there not being a reported Ct value for K3445002 with the DNA E primer. However, an estimated value was determined by using the Ct value for pGGA with the DNA E primer and the Ct value for K3445002 with the OriQ primer. The estimated value for K3445002 in vibrio was determined to be 6.73 copies of plasmid per genome. These values indicate that K3445002 is effective in both E. Coli and Vibrio natriegens, and there may even be a higher plasmid copy number in E. Coli with K3445002 in relation to the copy number for J04450. The dissociation curve was analyzed to verify that each reaction in the qPCR only produced one product. Taken together, these results show that we have improved the backbone of all parts (and J04450 specifically), resulting in a plasmid that is still capable of replication in E. coli, but now also available for use in Vibrio natriegens.
Team Amazonas_Brazil 2019:The RFP sinalization system in E. coli Nissle 1917
Characterization
Escherichia coli Nissle 1917 (EcN) is widely used as a probiotic since it’s discovery in the second half of the 19th century by Alfred Nissle1 and although many iGEM teams had worked with this bacteria, we couldn’t find any specific characterization about the manipulation of EcN. To improve the quality of our work and provide data for other teams or researchers that might want to work with this bacteria in their laboratory, we characterized EcN growth curve and it’s behavior when transformed by electroporation with the iGEM vector pSB1C3 with GFP and RFP.
E. coli Nissle 1917 strains were grown overnight in Lysogeny Broth (LB) containing ampicilin (100 µg/mL) at 37°C and 200 rpm. Cultures were diluted in fresh LB until achieve 0,1 OD with the corresponding antibiotic and transferred to a 96-well plate (50 µL/well). Samples were always made in triplicates and a blank of LB. During 8h the absorbance at OD600 and fluorescence (excitation 584 nm and emission 607 nm) were measured with intervals of 1 hour.
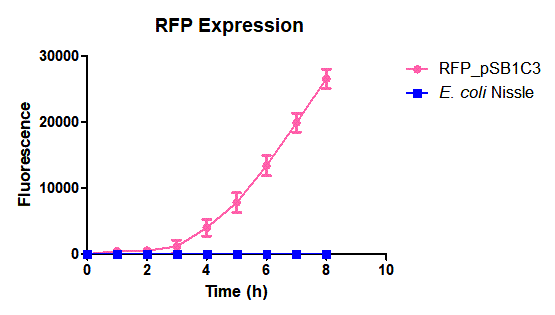
Fig. 1. Fluorescence values of E. coli Nissle 1917 curve(w/v)
Team NAU_CHINA 2019:The inhibitory effect of different concentrations of glucose in LB culture
Characterization
We used liquid M9 medium several times in our experiments, and the M9 medium contained a small amount of glucose (as a carbon source for bacteria). We used it to cultivate DH5α containing J04450 standard part; however, J04450 part contains a CAP-sensitive promoter, and glucose can inhibit its expression.
Since we were interested in the fact that glucose inhibits the expression of J04450, we explored the inhibitory effect of different concentrations of glucose on J04450 in LB culture. Add 50 ul of chloramphenicol to 5 bottles of LB medium and add IPTG to reach the concentration of 1%. Add 40% glucose to the concentration of 0,0.5%,2%,4%(w/v). Inoculate 1 ml of bacterial fluid and incubate at 37℃,150 rpm for every 0,8 h,16h,24h,28h,32h,36h. The phenotype was observed at 40h, and the change in fluorescence value was quantified by a microplate reader.
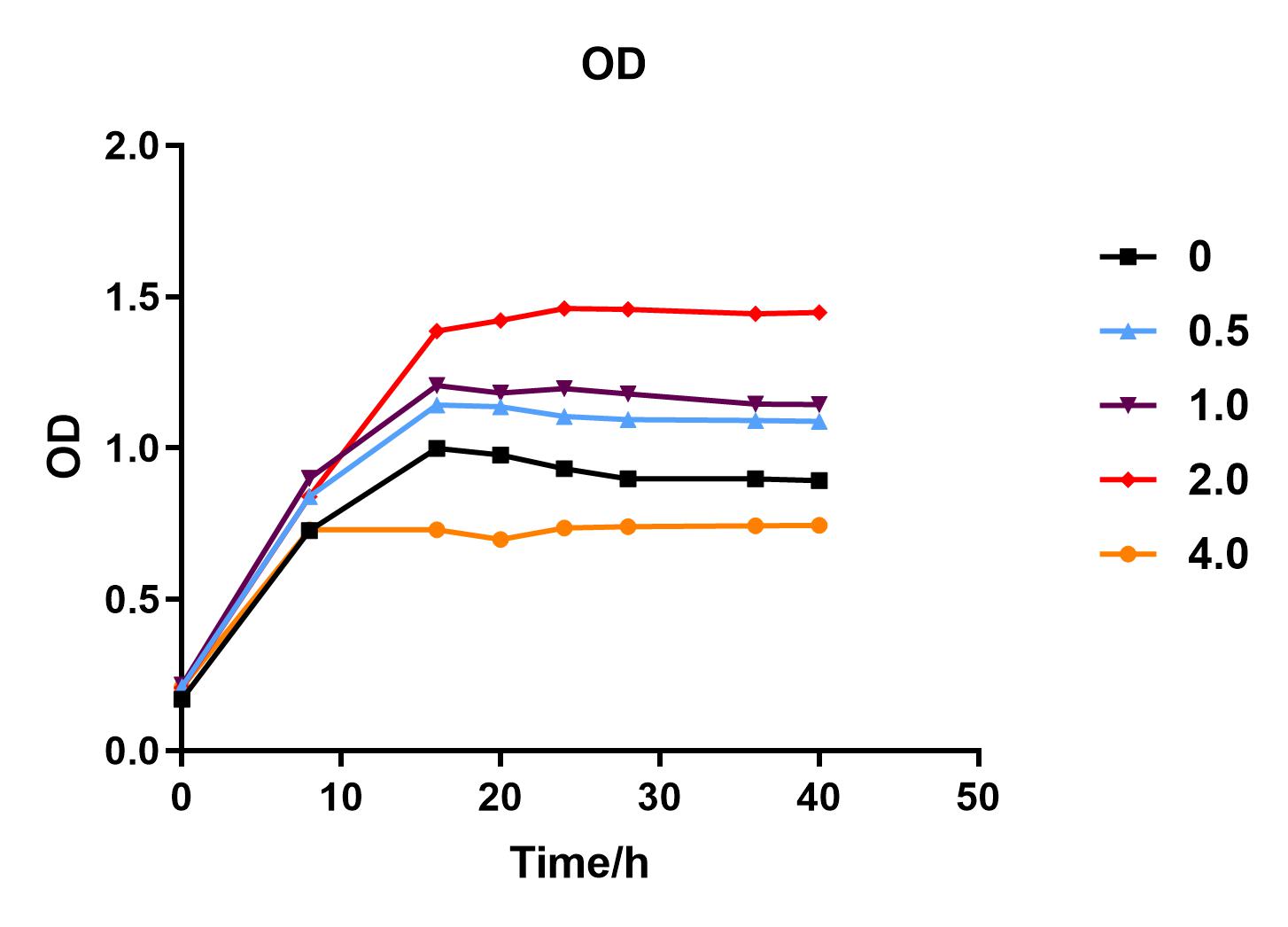
Fig. 1. OD600 values of bacteria at different glucose concentrations(w/v)
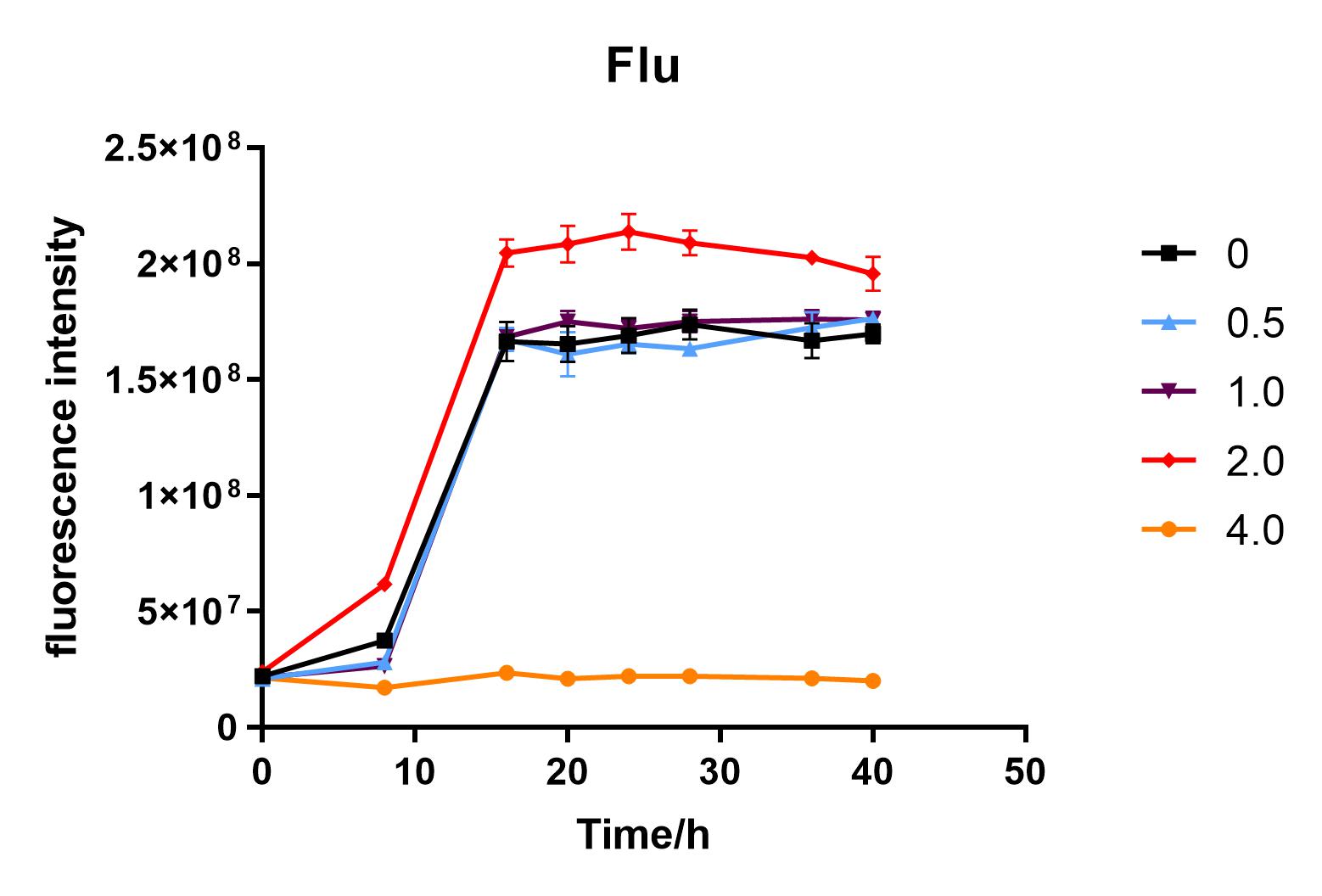
Fig. 2. Fluorescence intensity of bacteria at different glucose concentrations(w/v)
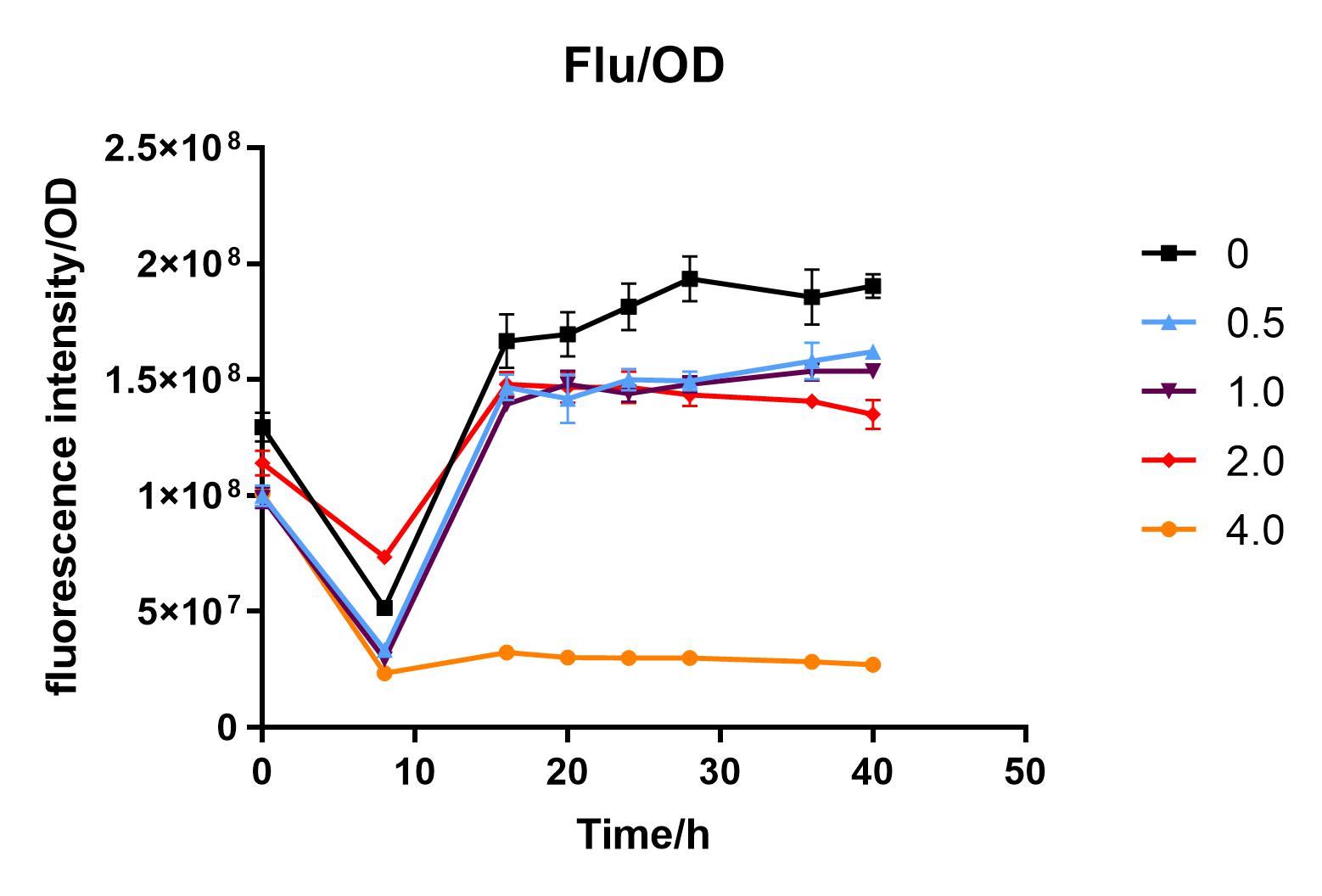
Fig. 3. Fluorescence intensity/OD value of bacteria at different glucose concentrations(w/v)
Analysis
We measured the OD value and fluorescence value of the sample by the microplate reader, and finally obtained the OD-time curve, fluorescence value-time curve, and fluorescence value/OD-time curve.
The data shows that the OD600 value of the cells decreases in the order of glucose concentration of 2.0%, 1.0%, 0.5%, 0, 4.0%, indicating that the proper amount of glucose can promote the growth of the cells, but the excess glucose has an inhibitory effect. The fluorescence intensity/OD value shows that different concentrations of glucose inhibited the expression of J04450, and the inhibition effect was the most significant at 4.0%.
Reference
[1]Mustea I , Muresian T . Crabtree effect in some bacterial cultures[J]. CANCER, 1967, 20(9):1499-1501.
Team ITB_Indonesia 2017: Red color dynamics of cloned Escherichia coli strains in LB broth

In Team ITB_Indonesia 2017
characterization, we found in normal growth/incubation condition (37 oC, LB agar) that
BBa_J04450-transformed Escherichia coli BL21 colony appear to need longer incubation time (>18
hours) until it clearly shows red color under natural light.
We then investigate whether this phenomenon is influenced by the strain, and we try if there are
lac repressor in the system that can be released by inducing the culture with IPTG, hence
increasing the expression of mRFP.
Experimental Design
We used three different strains of transformed E. coli (BL21, DH5alpha, and Top10) for this
study. They were incubated in LB broth, 37 oC, and sampled every 4 hours for 2 days to
determine the red color absorbance at 588 nm. The amount of IPTG added for respective treatment is 500
µM.
Result and Findings
- There are no significant differences of mRFP expression in different strains of E. coli (BL21, DH5alpha, Top10)
- There are no significant effects of mRFP increased expression after IPTG induction.
- The red color absorbance under 588 nm wavelength is recorded around 2.5-3 OD units.
- The broth become red in color under natural light around 16-20 hours of incubation time.


 ==Pictures==
==Pictures==


Team INSA-UPS France 2017 : usage in Vibrio harveyi strain engineered by conjugation
In the context of the iGEM INSA-UPS France project 2017, BBa_J04450 was tested in the Vibrio harveyi background. To the best of our knowledge, RFP has never been used in this strain. BBa_J04450 biobrick was cloned in a broad host range plasmid (pBBR1MCS-4) and conjugated into V. harveyi. The protocol of triparental mating can be found here. Its expression has been studied by fluoresence microscopy in Vibrio harveyi .

The microscopy results demonstrated the fonctional production of RFP in Vibrio harveyi, and hence, the functionality of part BBa_J04450 in this background.
IIT Madras 2016's Characterization


Experimentation
This BioBrick was used along with various GFP producing devices to understand the role of RBS and Promoter parts in giving rise to intrinsic noise in E. coli DH5alpha. Expression data for GFP and RFP proteins were obtained using flow cytometry (BD FACS Aria III) at 3hr, 6hr, 9hr and 12hr stage of growth along with cells expressing only GFP, only RFP and none. Cumulative intrinsic and extrinsic noise were measured using modified [http://2016.igem.org/Team:IIT-Madras/Model#Noise_in_Devices| Elowitz formula]. OD600 values for specific growth rate estimation were obtained using Spectrophotometer over an interval of an hour for 12 hours. Given specific growth rates are in it's logarithmic values. This BioBrick can be used to characterize noise and strength of complex devices by cloning this device with given device, which produces a different reporter protein. In graphs, we have R11-B32, R11-B34, J14-B3, J17-B34, R11-B30 and R11-B31 in pSB1A2 plasmid backbone.
[http://2010.igem.org/Team:Groningen Team Groningen 2010] reports the usage of this part as a cloning tool. When ligating any part, or part assembly, into any standard backbone that contains this part, the non-restricted and single-restricted backbones that self-circularize will produce red colonies on rich media plates (we use TY). These undesired transformants can than be avoided in the screening for the correct construct. With this method, the backbone desired for a new construct does not need to be purified from agarose gel to decrease the amount of undesired tranformants caused by ligation of the original part present in the backbone. The amount of incorrect transformants depends, of course, on the ratio of backbone (mixed with J04450) vs. BioBrick insert, the size of the BioBrick insert, and whether the insert is an assembly of two BioBricks. The images below show two ligations with different efficiencies.


Usage in Chromobacterium Violaceum
[http://2016.igem.org/Team:Tec-Monterrey Team Tec-Monterrey 2016] characterized the output of the part BBa_J04450 in a novel chassis, Chromobacterium Violaceum, as it produces a native purple pigment Violacein, we were curious whether RFP would be useful as a reporter gene. Furthermore, we characterized its expression under lac promoter. We did the transformation of C. Violaceum by a method that has not been reported yet, we made C. Violaceum competent cells with the protocol that is in our wiki, we concluded that the best O.D. for the heat shock transformation is 0.5 since it showed clearly better results than 0.4 or 0.6, we will continue to work in the transformation efficiency.
[Image: ]
]
H14Z1_Hangzhous 2018's Characterization
[http://2018.igem.org/Team:H14Z1_Hangzhou Team H14Z1_Hangzhou 2018]
Purpose
Previous experiments had shown that BBa_J04450-transformed E. coli BL21(DE3) would turn red color under natural light after 18 hours due to leaking expression. This part is useful for characterizing the expression of different proteins using the lacI sensitive promoter. For better application of this promoter in controlling protein expression, the effects of different hosts under various conditions were investigated systematically. The target hosts include E. coli BL21(DE3) and E. coli MC1061. The target culture conditions were temperature, inducer concentration and medium volume in the flask.
Experiment design
- Exploring the effect of temperatures on mRFP expression
1.Inoculate 5 ml LB with monocolony (BL21(DE3)/MC1061) from the plate. Grow the cells overnight at oC,200 rpm.
2.Dilute pre-culture 1:100 into 5ml LB (BL21(DE3)/MC1061) and cultivate at different temperature (26, 30, 34, 37 oC), 200 rpm, respectively.
3.Measure the fluorescence and OD600 every 4 hours. - Exploring the effect of medium volume on mRFP expression
1.Inoculate 5 ml LB with monocolony (BL21/MC1061) from the plate.Grow the cells overnight at 37oC, 200 rpm.
2.Dilute pre-culture 1:100 into 30ml, 50ml, 70ml LB (BL21(DE3)/MC1061) and cultivate at 37oC, 200 rpm.
3.Measure the fluorescence and OD600 every 2 hours. - Exploring the effect of concentration of IPTG on mRFP expression
1.Inoculate 5 ml LB with monocolony(BL21(DE3)/MC1061) from the plate. Grow the cells overnight at 37oC, 200 rpm.
2.Dilute pre-culture 1:100 into 5ml LB (BL21(DE3)/MC1061), cultivate at 37oC, 200 rpm. Protein expression are induced using 0.1, 0.3, 0.5, 0.8, 1.0 mM IPTG, respectively.
3.Measure the fluorescence and OD600 every 4 hours.
Results
- Exploring the effect of temperatures on mRFP expression
- Exploring the effect of effect of medium volume on mRFP expression
- Exploring the effect of concentration of IPTG on mRFP expression
There are some differences of growth rate at different temperatures for both E. coli (BL21(DE3), MC1061) (Fig.1). However, the differences of the fluorescence intensity at different temperature is much higher than those of the growth rate at various temperatures by using two E. coli cells (Fig 2). The fluorescence intensity followed same trend in two E. coli (BL21(DE3)/MC1061).The results indicated that the leaking expression was lower in low temperature (e.g.26 and 30oC) and higher in high temperature. Highest leak appeared in 34 oC. This finding is different from our expectation, where we expected that the highest leaking expression would be at 37 oC


The mRFP expression of two strains showed the same trend in different volume of medium in the flask. After 6 hours, the fluorescence intensity in 30 ml medium-containing flask is significantly higher than others, indicating that mRFP express fastest in this volume scale (Fig 3). This suggest that dissolved oxygen is an important factor which will affect the protein expressin and cell growth, since the lower medium volume in the same type flask will bring about higher level of dissolved oxygen in the medium.

Our results showed that significant fluorescence differences were not observed by using different IPTG induction (Fig 3). However, it is interesting that the fluorescence intensity of E. coli BL21(DE3) after IPTG induction are higher than those of E. coli MC1061, not similar to the trend in our previous experiment (Fig 2). It might be explained that the expression of mRFP can be accelerated by IPTG inductin in BL21(DE3) with regard to other E. coli hosts.

Pictures



SCAU-China 2018's improvement
The BBa_K2559005 is a amended eGFP coding part improved from BBa_l714891.
Usage and Biology
The part BBa_K2559005 has a sequence improvement on the basic part submitted by iGEM07_Peking (BBa_l714891) which encodes the SDY_eGFP. However, we found out a 16 bp nucleotides redundancy in the eGFP starting coding region in BBa_I714891, after checking the sequence of BBa_I714891 from NCBI. Therefore, we decided to delete the redundant 16 bp nucleotides in BBa_I714891 to amend the length of eGFP coding sequence. The amended eGFP coding biobrick is the BBa_K2559005. To test the function of BBa_K2559005, we designed a new E.coli expression vector containing our new part termed as BBa_K2559003under a strong E.coli endogenous promoter (PrplJ). Therefore, the amended eGFP in BBa_K2559005 was driven by PrplJ promoter, and expressed in DH10B. In addition, we also applied the BBa_K2559005 in the promoter intensity analysis of our other two new parts, the BBa_K2559004 and BBa_K2559011 which are relatively weaker E.coli endogenous promoters (PdapA and PcaiF) (Figure 1).


We summarized that our improvedpart, the amended eGFP coding biobrick BBa_K2559005 worked well in DH10B. We also hoped that our improvement on the BBa_I714891 can help their future applications by other groups in the future. However, it is difficult for us to perform additional experiments with BBa_K2559005 and BBa_I714891 due to the unavailable BBa_I714891.
To expand the application of BBa_K2559005, we searched theBBa_J04450 stored in registry and do another improvement in the BBa_J04450. The BBa_J04450 is a strong RFP expression vector in E.coli. As the main page of BBa_J04450 mentioned, the E.coli colonies with BBa_J04450 were in red color under normal light after about 18 hour culture on LB plate (Figure 2). We used the BBa_K2559005 to replace the RFP region in BBa_J04450, the modified part is BBa_K2559009. We transferred the BBa_K2559009 to DH5α by heat-shock, and found that the fluorescence signal can be observed under the UV (Figure 2).

So, we confirm that our improved part BBa_K2559005 can work in different E.coli expression system. We are also looking forward to more application of the BBa_K2559009! </html>
Grenoble-Alpes 2019 Characterization
The goal was to characterize the effect of cAMP on the promoter’s leakages and overall proteins expression
MAIN RESULTS
More informations here or in experience page.
DH5α : standard strain with endogenous adenylate cyclase
activity.
BTH101 : strain without endogenous adenylate cyclase activity
and so without production of cAMP. BTH101 strain is streptomycin
resistant.
BTH101 + adenylate cyclase (BTH101-Zip) to generate cAMP in the
strain.
To restore the adenylate cyclase activity we used two plasmids: one
containing pUT18-LeucineZipper (pUT18-LZ) and the other containing
pKT25-LeucineZipper (pKT25-LZ).
The Leucine Zipper’s (LZ) will
homodimerize and will bring T18 and T25 closer which will restore the
adenylate cyclase activity thus allowing the production of cAMP by
BTH101. Both T18 and T25 are under the control of an IPTG inducible
promoter (lactose promoter) to create an auto amplifier system.
BBa_J04450 was cloned in pKT25-LeucineZipper, and the bacteria was co-
transformed with both plasmid T25-LeucineZipper + BBa_J04450 and
T18-LeucineZipper.

It is apparent that without IPTG there is a visible difference between
strains that can produce cAMP (DH5α and BTH101-Zip) when compared with the BTH101 strain that do not produce any cAMP.
The DH5α red colonies reveal a substantial RFP expression as the result of the
leakage of the promoter.
BTH101 has white colonies indicating that no visible RFP was produced. This data
suggest that the promoter does not leak in that condition.
BTH101-Zip presents same red colonies than the DH5α strain.

RFP expression by BTH101 with or without cAMP shows that without cAMP, the lactose promoter has almost no leakage suggesting that the leakage of the lactose promoter is due to cAMP in absence of IPTG. When the cAMP production is restored the promoter is activated and the protein is expressed.
It is also interesting to note that with the BT101-Zip there are very little protein expression if no IPTG is added.
Tec-Chihuahua 2019 Characterization: RFP expression in E.Coli SHuffle strain
Introduction
E. coli SHuffle T7 Express®️ form New England BioLabs®️ was chosen for its ability to produce disulfide bonded proteins in its cytoplasm. However, no information was found in the Registry about the usage of part BBa_R0010 in SHuffle strains. In order to test this lacI regulated promoter in our chassis, we transformed part BBa_J04450, which expresses RFP under this promoter, into E. coli SHuffle T7 Express®️.Parts BBa_R0010 and BBa_J04450 were characterized. RFP expression was induced in different temperature conditions and IPTG concentration to determine the ideal conditions for the proper functionality of the promoter. Protein expression was analysed through an SDS-PAGE.
Experimentation
First, 3 μL of the 10 pg/μL vial of Part BBa_J04450 (RFP construct) from the Competent Cell Kit were transformed into chemically competent E. coli SHuffle T7 Express®️ cells. Transformed cells were grown on LB agar with chloramphenicol, for BBa_J04450 was located in the pSB1C3 backbone. Small, rounded, and red colonies grew on the surface of the agar.

Once the expression chassis was transformed, protein expression was induced. To do so, two tubes of 10 mL of liquid culture of untransformed E. coli SHuffle T7 Express®️ and six of SHuffle cells transformed with BBa_J04450 were prepared. Once the tubes reached an OD600 of 0.4, they were incubated for 5 hours under the conditions of temperature and IPTG concentration shown in the following table.
| Well | 37°C | Well | 30°C |
|---|---|---|---|
| Negative control | Untransformed control | Negative control | Untransformed control |
| Uninduced control (without IPTG) | Uninduced control (without IPTG) | ||
| |
Transformed cells, 0.2 mM IPTG | |
Transformed cells, 0.2 mM IPTG |
| |
Transformed cells, 0.4 mM IPTG | |
Transformed cells, 0.4 mM IPTG |
Induction temperatures were chosen based on commonly used conditions (37°C) and recommendations of the supplier (30°C). The uninduced control consists of transformed cells without IPTG which meant to serve as an indicator of leakiness in the promoter. IPTG concentrations were chosen based on conditions worked previously when using a T7 promoter (0.2 mM) and recommendations of the supplier (0.4 mM).
After 5 hours of incubation at 225 rpm, cells were centrifuged at 5000 rpm and 4°C for 7 minutes. The pellet was resuspended in a lysis solution with lysozyme and cells were lysed through multiple freeze/thaw cycles. The cell lysates were centrifuged at 21,380 g to separate the soluble protein fraction (supernatant) which was retrieved for its evaluation and quantification. 10 μL of each sample with 50 μg of total protein were mixed with 10 μL of SDS loading buffer and loaded into a 12% polyacrylamide gel. The gel was ran at 10 mA until the running front entered the separating gel; then, the current was increased to 15 mA.
Results
The following image shows the results obtained from the SDS-PAGE.

Two prominent bands can be appreciated in the last two columns, corresponding to induction conditions of 0.2 and 0.4 mM IPTG at 30°C, close to the 25 kDa band of the Precision Plus ProteinTM Dual Xtra Prestained Protein Standards. It was concluded that they correspond to RFP, a 26 kDa protein.
The band corresponding to RFP can be seen in each column with exception of the untransformed cells controls (negative controls). The uninduced controls (column 1 for 37°C and column 4 for 30°C) without IPTG were expected not to show bands if the promoter lacked leakiness; nevertheless, a faint band can be seen for both controls. Bands corresponding to 0.2 and 0.4 mM IPTG for both temperatures (columns 2 and 3 for 37°C and columns 5 and 6 for 30°C) show stronger bands compared to the uninduced control. Thus, the promoter is slightly leaky, but the addition of proper amounts of IPTG increases protein production. When comparing temperatures, incubation at 30°C clearly yields a higher amount of RFP, but yields similar amounts of expression between both concentrations.
Conclusion
We have demonstrated the functionality of promoter BBa_R0010 in E. coli SHuffle T7 Express®️ as well as ideal conditions for protein production by characterizing part BBa_J04450, an RFP construct. E. coli SHuffle T7 Express®️ produces higher amounts of protein at 30°C, and concentrations of 0.2 and 0.4 mM IPTG are enough to induce expression when using this lacI regulated promoter.
Team Lund 2019 charecterization
Aim
The aim of this characterization was to investigate the correlation between the different strains and how the varying amounts of salts and sugars in the media stimulate cell growth and protein expression. RFP was incorporated in the construct to compare the protein expression in various conditions. Three different E.coli strains were tested in three different complex media. The strains that were tested: E. coli TG1 E. coli BL21-DE3 E. coli Nissle 1917 (EcN) While the prepared media were: LB Media TB Media Enriched Complex Media The recipe for the three types of media can be found in our protocols.
Method
The plasmid from the registry was successfully transformed using heat shock to each of the mentioned strains. Three 1 L Erlenmeyer flasks containing 250mL of each media were prepared (9 flasks in total). Cultivation was performed at a temperature of 37 °C and 175 rotations per minute (rpm) , during which the growth was monitored spectrophotometrically. The protein expression could be detected optically from the color formation as well as with the use of Agilent Technologies Cary Eclipse Fluorescence Spectrophotometer at excitation wavelength of 532 nm and emission wavelength of 605 nm. For this experiment, a wavelength of 660 nm was used to measure optical cell density (OD) following the suggestion from iGEM headquarters.
The calibration curves for Rhodamine B (dye equivalent of RFP, obtained from Thermo Fisher Scientific - D1841) and for microbeads (equivalent of cells, provided by iGEM headquarters) were used to create a conversion factor for auxiliary fluorescence unit and Abs 660 nm. Two excitation wavelengths have been used 532 nm (recommended for RFP) and 570 nm (recommended for Rhodamine B). The protocol used to make the standard curves was the iGEM protocol “iGEM 2019 Plate Reader Fluorescence Calibration” for RFP.
Results
Standard curve
The emissions for these two excitation wavelengths can be seen in Figure 1 below.

Figure.1 The emission spectra for various concentrations of Rhodamine B (0 to 50μM): a) excitation at 532nm; b) excitation at 570nm.
One can notice that at excitation wavelength of 532 nm, a clear emission peak can be obtained approximately at 590 nm with the increase in signal intensity for each Rhodamine B concentration. While at excitation wavelength of 570 nm there are no clear peaks but the signal increases with the concentration of the fluorescent dye. Therefore, several standard curves have been plotted. The 532 nm/605 nm (ex./em.) was the one that was used for RFP, the 605 nm was a result of fluorescence shift due to presence of RFP in the cells. The 532 nm/590 nm was the setting to check the theoretical emission Rhodamine B at 590 nm using the same excitation as RFP. The last set of wavelengths 570 nm/590 nm was recommended by the manufacturer of Rhodamine B. The results are presented in Figure 2 below.

Figure 2. Rhodamine B standard curves showing mean fluorescence of four replicates of different concentrations of Rhodamine B (0 µM to 50 µM): a) excitation: 532nm, emission: 605nm; b) excitation: 532nm, emission: 590;c) excitation: 570, emission: 590
It can be clearly seen that in all cases there was an oversaturation of detector at concentrations of 12.5 μM and above. Therefore, it was decided that these concentrations should not be used for creation of standard curve and have been excluded. In case of 532/605, the noise is disturbing the measurement at lower concentrations but both 532/590 and 570/590 do not seem to have the same issues.

Figure 3. Rhodamine B standard curves showing mean fluorescence of four replicates of different concentrations of Rhodamine B (0 µM to 6.25 µM – the higher values were excluded due to oversaturation):a) excitation: 532 nm, emission: 605 nm; b) excitation: 532 nm, emission: 590 nm;c) excitation: 570 nm, emission: 590 nm
It can be seen that all the curves show similar linearity. The data from the curves was used to determine a unit scaling factor (μM Rhodamine B/ fluorescence a.u.) that can be found in Table 1, below.
Table 1. The mean μM of Rhodamine B per fluorescence auxiliary unit (a.u.)

Since all the standard curves have similar linearity but the scaling factors are significantly different, it was decided to choose the manufacturers recommendation and use the date from 570nm excitation and 590nm emission, that is 0.23 μM Rhodamine B/a.u.. It was hence used to convert the fluorescence auxiliary units in the characterisation experiment.
The calibration curve for the particles can be seen in Figure 4. below.

Figure 4. The calibration curve for absorbance of various concentrations silica particles at 660 nm.
The calculated unit conversion factor was 1,23E+09 particles/Abs660 and it was used to convert units in characterization experiments.
The RFP/Rhodamine B Fluorescence Standard Spreadsheets found here for all excitation/emission can be found here:
Characterization results
Figure 5a below shows the growth of EcN in different media in a fraction of time. Enriched Complex Media appears to be the most suitable for the growth of EcN. The lag phase is shorter compared to the other two media, while the logarithmic phase is longer. In TB Media the lag phase is extended, and the growth is not as high as before but still exhibits a clear logarithmic phase. In sharp contrast to that, the lag phase in LB Media is almost double and the exponential phase is quite brief. These results are expected since the media most enriched with salts and sugars are the media with the highest growth rate. The same applies to all strains (Figure 5b and Figure 5c).

Figure 5. The growth of various E. coli strains expressing RFP in different media: a) EcN in LB (blue), TB (orange), EM (grey). b) E. coli BL21-DE3 in LB (blue), TB (orange), EM (grey) c) E. coli TG1 in LB (blue), TB (orange), EM (grey). The OD was converted to equivalent particle count/100μL.
Figure 6 below shows the expression of RFP in different media. In EcN (Figure 6a), the expression of RFP for TB and Enriched Media are comparable, whereas the RFP production in LB is significantly lower. For E. coli BL21-DE3 (Figure 6b), however, the expression levels in TB and LB Media are similar and in Enriched media the RFP production was considerably higher. Finally, in E. coli TG1 (Figure 6c) contrary to the other two strains the Enriched Media gives the lowest RFP levels. The expression in LB media is only slightly higher than in Enriched media. The protein expression in TB Media, however, is notably higher than the other two media, and in comparison to the other strains in the same media. In all three strains, the production of the protein was initiated in LB Media but it could not reach as high values as in the more fortified media, which was expected.

Figure 6. The expression of RFP in various E. coli strains in various cultivation media: a) EcN in LB (blue), TB (orange), EM (grey). b) E. coli BL21-DE3 EcN in LB (blue), TB (orange), EM (grey) c) E. coli TG1 EcN in LB (blue), TB (orange), EM (grey). The OD was converted to equivalent particle count/100μL.
Figure 7 below shows the expression of RFP (converted to Rhodamine B equivalents) per OD (converted to particle/100uL). When it comes to growth, all strains prefered Enriched media. However, it can be noted that each strain has a different preference regarding the RFP expression. EcN produces the most RFP per cell in TB media, BL21 in LB media while in TG1 the expression is favored in both LB and TB media.

Figure 7. The production of RFP per particle count (OD equivalent) for each strain in various media at the end of cultivation: LB (blue), TB (orange), EM (grey).
Figure 8 displays growth of the three strains of E.coli during the characterization of RFP. Overall, the darkest red color was from cells grown in TB media. This was where the protein expression was higher. The other two media, complex Enriched media and LB media both gave a quite pale pink color and low protein expression, with one exception of the BL21-DE3 cells which gave a dark red color and high protein expression. For BL21-DE3 cells, the complex enriched media showed the darkest color. For TG1 cells the TB media gave the darkest color. For EcN cells the TB media also gave the darkest color. These correspond to the results presented in Figure 7.

Figure 8. Growth of three strains of E.coli cells transformed with RFP plasmid BBa_J04450. The cells were grown in different media. The three flasks on the left were grown in TB media, and the strains were from left to right; BL21-DE3, TG1, EcN. The three flasks in the middle were grown in complex Enriched media, and the strains were from left to right; BL21-DE3, TG1, EcN. The three flasks on the right were grown in LB media, and the strains were from left to right; BL21-DE3, TG1, EcN.
Usage and Biology
Using cAMP depleted strains and T18-leucineZipper/T25-leucineZipper under the control of a lactose promoter is a good alternative to produce inducible proteins without leakage
Team BUCT_China 2019
Experimental Design
1.The target gene was obtained by PCR cloning.
2. Digest the gene and plasmid pSB1C3 with enzymes Dpn,IEcoR,Pst I.
3.Ligate the digested gene fragment and plasmid pSB1C3 with T4 enzyme.
4.Transfer the ligation product to Trans 10 by thermal conversion.
5.Plate the transformed strain and culture at 37℃ for 12h.
6.Pick a single colony from the plate to make a bacterial solution.The cells were cultured separately in 24 tubes, and every tube was measured the absorbance in order for each 1 hour.
Results
The growth of the plate colonies was observed as shown in the Figure a. We could see that an appropriate amount of colonies grew on the plate, indicating that the target gene fragment was successfully cloned into Trans 10.

We quantitatively described the growth curve of Trans10 single colony in medium.(Figure b) According to the growth rate constant of the bacteria, we could divide 24h into three different growth phases:0~2h is lag phase; 2~ 15h is an exponential phase; 15~ 24 is a stationary phase.

Newcastle 2019 characterisation
The Newcastle iGEM team aimed to develop a suite of biosensors using fluorescence proteins as a reporter. Our goal was to investigated if we can measure fluorescence level correctly when fluorescence proteins are combined. We investigate whether mixing cultures of cells expressing different fluorescent proteins would affect the fluorescence intensity of the individual fluorescent proteins. The fluorescent proteins we chose were sfGFP (BBa_K515105) and mCherry (BBa_J04450).
We obtained sfGFP and mCherry into pSB1AT3 from iGEM distribution kits. The resuspended DNA was transformed into E. coli DH5 alpha cells. Cells were inoculated in LB media and grown to an optical density 600 (OD600) of 0.6. Half of the culture was diluted to OD600 0.3 and the individual fluorescence levels were measured at: sfGFP - excitation 480nm and emission 507 nm and mCherry – excitation 580 nm, emission 610 nm. The sfGFP and mCherry cultures at OD600 0.6 were mixed together in equal amounts and the fluorescence level of the combined mixture was measured in a BioTek Synergy H1 Microplate Reader with a gain of 100.
The sfGFP and mCherry cultures at OD600 0.6 were mixed together in equal amounts and the fluorescence level of the combined mixture was measured. Theoretically, the OD600 of each fluorescence protein in the mixture will be 0.3. By comparing the individual fluorescence level at OD600 of 0.3 to the fluorescence level of the equivalent fluorescence protein minus the opposing fluorescent protein, the effect of mixing fluorescent proteins can be observed.
The emission spectra for sfGFP ranges from 469 nm to 628 nm and the emission spectra for mCherry ranges from 551 nm to 800 nm. There is an overlap of 77 nm in the emission spectra of the two fluorescent proteins. This small overlap suggests that there will not be much effect on the fluorescence intensity of the individual fluorescent proteins as the overlap will be at an emission with a low percentage emission.

Figure 1. A) Bar chart showing the mean fluorescence intensity of E. coli DH5 alpha cells expressing sfGFP individually at an OD600 of 0.3 with standard deviations compared to the mean fluorescence intensity when mixed with cells expressing the fluorescent protein mCherry Excitation 480 nm, emission 507. B) Bar chart showing the mean fluorescence intensity of E. coli DH5 alpha cells expressing mCherry individually at an OD600 of 0.3 with standard deviations compared to the mean fluorescence intensity when mixed with cells expressing the fluorescent protein sfGFP. Excitation 580 nm, emission 610 nm.
The results of mixing sfGFP and mCherry at sfGFP excitation and emission (Figure 1A) showed that there was no significant difference in fluorescence intensity of sfGFP when mixed with mCherry (Figure 13A). The mean fluorescence of sfGFP individually was 168008 ± 2503.4 compared to the mean fluorescence of sfGFP mixed with mCherry at 172678.2 ± 6826.9. Similarly, at mCherry excitation and emission, similar results were observed and there was no significant difference in fluorescence intensity (Figure 1B). The mean fluorescence of mCherry individually was 4524.8 ± 5.1 compared to the mean fluorescence of mCherry mixed with sfGFP at 4693.6 ± 162.5.
In conclusion, mixing of sfGFP and mCherry did not significantly alter the fluorescence intensity of each individual fluorescent protein. It can be concluded that despite the mixing of fluorescent proteins, when measuring fluorescence intensity, it can be confidently assumed that the results for each fluorescent protein is accurate.
For more detail see - https://2019.igem.org/Team:Newcastle/Results/bronzecharacterisation
Team Aalto-Helsinki 2019 characterization: Effect of antibiotic concentration on fluorescence intensity
Aim
The aim was to characterize the RFP coding device (BBa_J04450) by measuring the effect of a range of different chloramphenicol concentrations on fluorescence intensity in Escherichia coli DH5a.
Method
For the experiment, a DH5a colony harboring the BBa_J04450 was used to inoculate 20 ml of LB medium supplemented with 25 ug/ml chloramphenicol. The culture was grown for 24 h at +37 C with 250 rpm shaking. The experiment was carried out on a 96-well plate with 90 ul of LB+cam and 90 ul of cell culture in each well. The following chloramphenicol concentrations were tested: 12,5 mM; 17,5 mM; 25 mM; 32,5 mM; 62,5 mM; 87,5 mM. Concentrations represent final concentrations in each well after addition of cell culture.
Measurement was carried out with Synergy H1 microplate reader (Biotek). Excitation read was set to 556 nm and emission to 586 nm. OD600 was measured from each well. Optics were read from the bottom of each well. Temperature was set at 37 C (+ preheat) and the plate was under continuous shaking with 230 rpm. Fluorescence and absorbance readings were measured every 10 min over 8 h. Mean fluorescence and OD600 values were calculated from three replicates at each time point. Fluorescence values were normalized with OD600 values so that differences in cell growth would be considered. As a reference for this method, we used a protocol kindly provided by Christopher Jonkergouw from Aalto University. The protocol can be found in our WIKI page under Protocols (18. Fluorescence expression in Microtitre).
Results
OD600 values for each tested antibiotic concentration are presented in Figure 1. Absolute fluorescence values are presented in Figure 2, and normalized fluorescence values in Figure 3.

Initial OD600 levels were between 0.1-1. Cell density of cultures supplemented with lowest antibiotic concentration (12,5) increased from 1.0 to 1.3 during 8 h incubation. Cell density of cultures with higher antibiotic concentrations increased slightly less, up to 1.1 approximately.

Fluorescence values were highest in cell cultures supplemented with lowest antibiotic concentrations (12,5 mM), following a similar pattern to cell density. Second highest fluorescence values were observed with cells supplemented with 25 mM, followed by 62,5 mM concentrations.

Normalized fluorescence values increased in all chloramphenicol concentrations we tested. Fastest increase was observed with the lowest antibiotic concentration. After 8 hours of incubation, highest antibiotic concentration showed the highest fluorescence readings. This is probably because cell density in 87,5 mM cultures increased after 6,5 h incubation.
In conclusion
It was shown that different chloramphenicol concentrations have an effect on fluorescence intensity. Lower chloramphenicol concentration (12,5 mM) which is typically applied could maintain RFP expression in DH5 for 8 h. It was also shown that cells were able to grow and express RFP with chloramphenicol concentration of 87,5, which is 3,5 times higher than the recommended concentration (25 mM). Cells supplemented with 17,5 and 32,5 mM concentrations had lowest expression of RFP of the range of concentrations studied here.
Team Botchan_Lab_tokyo 2019 characterization
description
We transformed the plasmid which RFP is inserted in psB1C3 into DH5α, and tested radiation tolerance. The following graph shows the result. About protocol, please refer to 3.2 RFP in the protocol of our that's page.
Experimental Design
Gamma-irradiation and cell survival assay
We transformed DH5α with the plasmid which RFP is inserted in psB1C3 and tested radiation tolerance.
・At first the cells are cultured in liquid medium and the medium is replaced with Butterfield‘s Phosphate Buffer (BPB). This is because when the gamma ray is irradiated, the complex culture medium might affect the cell and it might disturb the results. So it‘s necessary to replace the complex and unknown factor before gamma-irradiation.
・After replacement with BPB, irradiate a certain amount of gamma rays (0, 10, 50, 100 Gy*) and spread the solution on the plate. Incubate them at 30 ℃.
・Finally, count the number of colonies that appeared a few days later (48-72 hours) and calculate the average Colony Forming Unit (CFU/ mL, equal to the viable cell concentration). The average CFU / mL of an irradiated sample (N) was divided by the average CFU / mL of a non-irradiated sample (N0) to produce the survival rate (N0/N).
- Gy = J/kg, the SI unit of energy absorbed from ionizing radiation.
Result
survival rate of E. coli (have plasmid RFP(BBa_J04450)inserted)

Figure1. The viability of E. coli DH5α against gamma irradiation
E. coli showed a significant decrease in viability between 50 Gy and 100 Gy versus 10 Gy. From this result, we concluded that E. coli is highly sensitive to gamma irradiation when it is irradiated with 50 and more doses of Gy (As for 0 Gy, an irradiated sample was as same as a non-irradiated group, so the survival rate was calculated as always 1).
conclusion
This graph shows that the number of cells is significantly reduced with respect to the intensity of radiation. That is, it was shown that E. coli (DH5α) having a plasmid inserted with RFP has no radiation resistance.
Contribution: Gunma 2021
Summary
Considering productivity of the expressed protein, it is more important, particularly in a real world, to exactly know how much time it takes to obtain as large an amount of the expressed protein as possible per a given volume of the culture media. To learn more about the characteristics of the plasmid expressing RFP proteins, we did experiments as below.
Experiments
The plasmid BBa_J04450 was transformed into DH5α. The transformant as well as the control was grown at 37 ℃ in LB media in a shaking incubator (130 rpm). We measured OD600 of each sample, and at the same time visually confirmed color of the culture media, since RFP is a red fluorescent protein.
Results and discussion
The results showed that after 16 hours, the number of cells was the largest and the medium became even deeper in red, indicating that 16 hours are at least required for productivity of the expressed protein in this conditions. It is quite likely that the promoter activity of the plasmid is most induced in the stationary phase.

improvement: NAU-CHINA 2021
(more details see our part registry)
Bacterial 5’ untranslated regions as an important part of mRNA (5’ UTR), it involves in a complex regulation of gene expression; however, the exact sequence features contributing to gene regulation are not yet fully understood. But many researches have proved that 5’ UTR plays a central role in gene expression and protein production. At the DNA level, it involves in transcript formation for the interplay between promoter and initially transcribed sequences (ITS). But, at mRNA level, it influences transcript stability and translation because of secondary structure formation and so on. Therefor 5’ UTR is has a crucial contribution to the maintenance of a fine balance between transcription, transcript stability and translation. In the work of Simone Balzer Le, Ingerid Onsager et al, they design a novel 5’UTR-Dual UTR, utilizing the transcriptional and translational characteristics of 5’UTRs in a single expression cassette, which will improve protein expression greatly.
The Dual UTR contains two different UTRs which separated by a spacer region (Fig1). The 5’ UTR proximal to the promoter originates from the transcriptional screening(Tr-UTR) which affects transcript formation, while the second 5’ UTR is identified from the translational screening (Tn-UTR) which influences transcript stability and translation. And the spacer region provides enough space for physical separation of mutations affecting transcription and translation. By using the Dual UTR, we can improve the protein expression greatly.

Figure.1 Dual UTR(BBa_K3735020)
Our design:
After learning about the Dual UTR, we want to utilize it to improve an existing measurement part so that subsequent researchers can detect fluorescence more efficiently and accurately. Then we chose the BBa_J04450 which expresses mRFP under the induction of IPTG. We intend to add the Dual UTR between mRFP(BBa_E1010) and lac promoter(BBa_R0010) to improve its expression. Thereupon, we construct the following part(BBa_K3735666).

Fig.2 Improved mRFP expression element(BBa_K3735666)
Characterization:
We constructed the expression plasmid(BBa_K3735666) by using homologous recombination, which with Dual UTR sequence inserted. Then the recombinant plasmid was transformed into E.coli BL21(DE3) competent cells and grown overnight. Then the single colony(BBa_K3735666) and control(BBa_J04450) were used to inoculate in 50ml LB medium containing chloramphenicol and cultured to the same state. We cultured them under the induction of IPTG, and measured OD600 and fluorescence intensity per 2 hours.

Fig.3 Plasmid map(A: Improved part (BBa_K3735666); B:Original part(BBa_J04450))
Results:
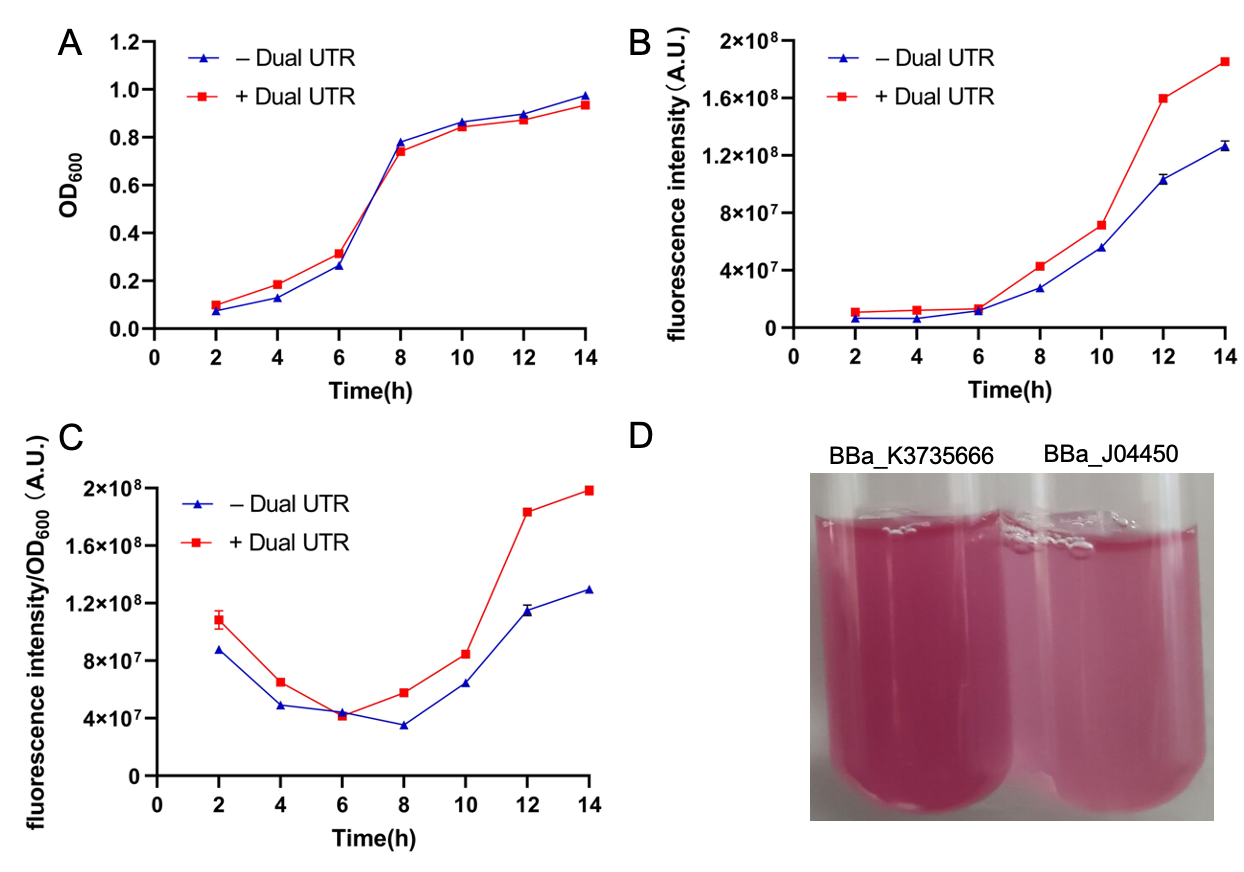
Fig.4 The results of improvement. A: the value of OD600 of BBa_J04450 and BBa_K3735666; B: the fluorescence intensity of BBa_J04450 and BBa_K3735666; C: the fluorescence intensity/OD600 of BBa_J04450 and BBa_K3735666
We can see that the fluorescence intensity of the improved part(BBa_K3735666) is significantly higher than that of the control group(BBa_J04450). Increasing the expression of fluorescence intensity can amplify weaker signals. At the same time, our method is also suitable for improving the expression intensity of other parts.
(more details see our part registry)
Improvement: NAU-CHINA 2022
Improvement
The Ssr tag exists in bacteria extensively, serveing as a central feature of protein-quality control system. When protein translation stalls, the ribosome-rescue system mediates the addition of the sequence AANDENYALAA, an Ssr tag, to the C-terminus of the nascent polypeptide. The peptide sequence will lead the modified protein to degradation. In addition, Tobacco Etch Virus (TEV) protease is a site-specific protease which has the ability to recognize the amino acid sequence consisting of ENLYFQS and cleave it between Q and S. Taking advantage of the characteristics of TEV protease and Ssr tag, we can integrate a logic operation to decrease the leakiness of parts.
Plac promoter is a widely used promoter in induced expression of proteins. The gene products of lacI is a repressor for lac operon which can bind to lac operator, then block the RNA polymerase from binding to the Plac promoter and switching on transcription. When induced by IPTG, LacI can be removed from the site so that the transcription of downstream gene will be turned on. BBa_J04450 is a Red Florescence Protein (RFP) expression device driven by Plac promoter. When it works in E.coli , a quantity of RFP will express without IPTG due to the low expression level of LacI in the chassis. JNFLS-2020 has managed to reduce the leakiness by linking lacI and RFP to the downstream of Plac. This year, NAU-CHINA tried to build an anti-leakage device on BBa_J04450 which can be stretched to extensive inducible promoter.
The improvement of NAU-CHINA this year is to integrate TEV protease and Ssr tag to construct BBa_K4164020 in order to control the degradation of mRFP and resolve the leakage problem of BBa_J04450.
TEV cleavage site (ENLYFQS) and Ssr tag (AANDENYAAV) are inserted into the C-terminus of RFP in turn (Fig.1). Meanwhile, the TEV protease is expressed also driven by Plac. The Ssr tag can lead to the degradation of RFP expressed without IPTG. When induced by IPTG, the TEV protease can be inductively expressed, and cleave specifically at the cleavage site, which results in the loss of Ssr tag in RFP, and RFP lost the degradation tag will no longer be degraded and begin to work.

Figure 1. Improvement of mRFP expression device.
We used pET29a(+) as the vector, which is capable of expressing LacI to conduct BBa_K4164020. Additionnaly, we constructed BBa_J04450, BBa_K4164021 and BBa_K4164022 (Fig.2) in pET29a(+) as control groups. These plasmids were transformed into E.coli BL21 respectively and grown in LB medium at 37℃ until the optical density at 600nm (OD600) reached 0.6. After that, the bacteria culture fluid was incubated in 50mL LB medium with or without IPTG induction at 37℃ for 12 hours, and measured the OD600 and fluorescence intensity (excitation 535 nm and emission 605 nm) per hour.

Figure 2. a.Improved part BBa_K4164020; b. Original part BBa_J04450; c. Control part 1 BBa_K4164021; d. Control part 2 BBa_K4164022
We find that the leakage level of BBa_K4164020 is lower compared with BBa_J04450 and BBa_K4164021. What’s more, due to the absence of TEV protease, the fluorescence intensity of BBa_K4164022 is poor regardless of whether there is IPTG induction. However, the relative fluorescence intensity of BBa_K4164020 and BBa_J04450 are similar in the condition of IPTG induction, which suggests the Ssr tag has little influence on the expression of RFP under normal circumstances.

Figure 3. the result of improvement.
a. the variation of fluorescence intensity/OD of BBa_K4164020, BBa_J04450, BBa_K4164021 and BBa_K4164022 cultivated with or without IPTG induction;
b. the fluorescence intensity/OD of BBa_K4164020, BBa_J04450 and BBa_K4164021 cultivated without IPTG induction;
c. the fluorescence intensity of BBa_K4164020, BBa_J04450, BBa_K4164021, BBa_K4164022 and Control cultivated with IPTG after 12 hours;
d. the fluorescence intensity of BBa_K4164020, BBa_J04450, BBa_K4164021, BBa_K4164022 and Control cultivated without IPTG after 12 hours.
Contribution: NYCU_Formosa 2022
Color mixing model
To establish our color mixing model, we measured the fluorescent intensity to time relationship of Part: BBa_J04450(rfp) and Part: BBa_K741002(gfp). With these parameters, we created a model for future teams to deduce the ratio of red and green for their desired color. This simulation shows that different RGB values lead to different color results. What’s more important is that the changing degree of red and green values is based on our protein expression experiments. Thus, according to their desired color, future teams are able to speculate the specific incubation time for E.coli transformed with RFP and GFP genes.
With experimental verification, we have mixed different concentrations of RFP and GFP in fixed value of 200μL, which includes ratio 1:1, 5:3, 2:1, 3:1, 4:1, 9:1, 1:9, 4:1, 3:1, 2:1.

Figure 1. RFP : Flourscecnce Intensity of RFP over time

Figure 2. GFP : Flourscecnce Intensity of GFP over time

Figure 3. Different combinations of RFP and GFP

Figure 4. Combinations of RFP and GFP of 1:9 and 9:1.

Figure 5. Combinations of RFP and GFP of 1:5 and 5:1.

Figure 6. Combinations of RFP and GFP of 1:4 and 4:1.

Figure 7. Combinations of RFP and GFP of 1:3 and 3:1.

Figure 8. Combinations of RFP and GFP of 1:2 and 2:1.

Figure 9. Color Mixing Model 1.

Figure 10. Color Mixing Model 2.
Team NAU-CHINA 2023 contribution: Effect of mRFP expression on the cell abundance measurements
Purpose & Design
In our contribution section, we decided to delve deeper into the investigation of the commonly used reporter gene, Red Fluorescent Protein (RFP). Red Fluorescent Proteins (RFPs) possess a strong absorption of light at the 600 nm wavelength, leading to a misrepresentation of cell optical density and consequently an underestimation of single-cell fluorescence. Thus, we selected BBa_J04450 from the iGEM part repository and used pSB1C3 as the vector to introduce it into Escherichia coli DH5α for expression, aiming to explore the influence of different light wavelengths on the deviation in measuring Red Fluorescent Protein (RFP) abundance in cells.

Fig. 1 The Plasmid Map of pSB1C3-J04450.
Experiment
1. We activated BBa_J04450 obtained from the kit and transformed it into E. coli DH5α, followed by overnight incubation at 37℃. The subsequent day, we chose 4 colonies and cultured them in 5 mL LB medium for approximately 12 hours. This genetic segment was expressed within the plasmid pSB1C3; consequently, both our LB agar plates and culture media were supplemented with chloramphenicol (50 μg/mL).
2. Used XbaⅠ and SpeⅠ enzymes to construct the empty vector pSB1C3.

Fig. 1 The Plasmid Map of pSB1C3.
3. The pSB1C3 plasmid was transformed into E. coli DH5α, followed by overnight incubation at 37℃. The next day, four colonies were selected and cultured in 5 mL LB medium for approximately 12 hours.
4. We prepared six fresh 50 mL LB media. Subsequently, we introduced overnight cultures of E. coli carrying pSB1C3-J04450 and pSB1C3 , respectively, into these media. The initial three media were inoculated with pSB1C3 E. coli, while the latter were pSB1C3-J04450 E. coli. These cultures were cultivated for sixteen hours. During the sampling process, we transferred 200 μL cultured samples into Costar 96 Flat Bottom Polystyrene Cat with black and transparent bottoms. Notably, each sample was subjected to three replicates within both the black and transparent groups.
5. After sampling, we promptly measured the data using an microplate reader (Tecan Spark) and conducted subsequent analysis.
Results
Among commonly used fluorescent proteins, the excitation wavelength for mRFP is situated around 585 nm (Fig. 2). Consequently, it exhibits a prominent absorption peak at 600 nm (Fig. 2), a standard wavelength used for measuring bacterial culture optical density (OD). This phenomenon leads to a significant overestimation of data when quantifying the abundance of bacteria expressing red fluorescent protein.

Fig. 2 Absorption spectra of mRFP1.
Initially, we measured the fluorescence levels of E. coli. Notably, the E. coli carrying pSB1C3-J04450 exhibited apronounced shift towards red fluorescence in the later stages. In the early stages, E. coli carrying pSB1C3-J04450 displayed fluorescence levels similar to those without it. However, after 12 hours, a substantial increase was observed, significantly surpassing the fluorescence levels of the E. coli carrying pSB1C3 (Fig. 3). This indicates the density of our experimental strain, which exhibits a distinct disparity compared to E. coli carrying pSB1C3.

Fig. 3 The Fluorescence of each culture.
We measured the OD values of the bacteria, revealing distinct differences between Escherichia coli strains carrying pSB1C3-J04450 and those carrying pSB1C3 at various wavelengths. Specifically, data obtained under 585 nm wavelength exhibited the most significant disparities between the two E. coli strains, while data acquired under 700 nm wavelength displayed less obvious distinctions (Fig. 4a-d). The increased disparity at 585 nm can be attributed to the absorption peak of mRFP occurring near this wavelength, thus making the data differences between the two strains more pronounced. Thus, the influence of RFP can be disregarded at the two longer wavelengths.
Furthermore, to gain further insight into the impact of RFP on E. coli OD values, we conducted deviation analyses relative to the baseline at 700 nm and performed linear function fitting for the data obtained at the other three wavelengths. It is evident that the data acquired at 660 nm exhibited minimal deviation, while the other two shorter wavelengths displayed a noticeable upward trend (Fig. 4E). At the 12-hour mark, the OD values obtained at 600 nm were nearly 15% higher than those unaffected by RFP. This illustrates the impact of mRFP expression on the measurement of bacterial density under wavelength at 600 nm. This serves as a valuable reference for future teams.

Fig. 4 The ODabs and Deviation Analyses a-d ODabs measurements were taken at wavelengths of 585 nm, 600 nm, 660 nm, and 700 nm. Due to the absorption peak of RFP at 585 nm, E. coli carrying pSB1C3-J04450 exhibited significantly higher data values at both 585 nm and 600 nm wavelengths compared to those carrying pSB1C3. However, data at 660 nm and 700 nm showed relatively minor differences. e: Using 700 nm as reference, bias analysis and function fitting were conducted for the data obtained at the other three wavelengths. In comparison to OD at 660 nm, the OD at 585 nm and 600 nm displayed a notable positive correlation, highlighting the significant impact of RFP expression on bacterial concentration measurements at 585 nm and 600 nm.