
Difference between revisions of "Part:BBa I719005"
Phototroph (Talk | contribs) (→St_Andrews 2019 characterization) |
|||
| (65 intermediate revisions by 9 users not shown) | |||
| Line 42: | Line 42: | ||
=St_Andrews 2019 characterization= | =St_Andrews 2019 characterization= | ||
| − | We characterised | + | <html> |
| + | <p>We characterised this part using an <i>in vitro</i> T7 polymerase transcription system, measuring the concentration of Firefly Luciferase RNA transcript at 3 different temperatures (30°C, 37°C, 45°C), produced over time. Detail of the protocol can be found on our <a href="https://2019.igem.org/Team:St_Andrews/Experiments">Experiments</a>page.</p> | ||
| + | <p>We fitted our results using a beta regression in R, with p << 0.01 for all models. Our results are plotted below. The regressed model is shown by a bold line and a 95% confidence interval is bounded by two dashed lines. </p> | ||
| + | <div class="col-sm-12" align="center"> | ||
| + | <img src="https://2019.igem.org/wiki/images/f/fd/T--St_Andrews--RNA_confidence_intervals.png" alt="Graph of RNA production over time" style="width:75%;"></img> | ||
| + | <p><font size=-2><i><b>RNA Production over time:</b> Concentration of Firefly Luciferase RNA transcript produced using T7 Polymerase and BBa_I719005 in vitro over time. Models of RNA as a function of time are shown in bold line, with 95% confidence intervals for each model bounded by two dashed lines. </i></font></p> | ||
| + | </div> | ||
| + | <p>Our results show a highest expression rate of transcript at 37<sup>o</sup>C when using T7 polymerase <i>in vitro</i>.</p> | ||
| − | + | </html> | |
| − | + | ||
=ZJU-China 2019's improvement= | =ZJU-China 2019's improvement= | ||
==I Overview== | ==I Overview== | ||
This year team ZJU-China has upgraded four modified T7 promoters with increased strength (<partinfo>K2965011</partinfo>, <partinfo>K2965012</partinfo>, <partinfo>K2965013</partinfo>, <partinfo>K2965014</partinfo>). | This year team ZJU-China has upgraded four modified T7 promoters with increased strength (<partinfo>K2965011</partinfo>, <partinfo>K2965012</partinfo>, <partinfo>K2965013</partinfo>, <partinfo>K2965014</partinfo>). | ||
| − | With the help of previous research, we carefully chose the site which would be mutated by PCR. These sites distribute in the range from -3 to +6. We mutated these sites by adding variable bases to primers (taatacgactcactatagggaga → taatacgactcacWNNNgSRRNN), and screened for stronger mutants. | + | With the help of previous research[1-2], we carefully chose the site which would be mutated by PCR. These sites distribute in the range from -3 to +6. We mutated these sites by adding variable bases to primers (taatacgactcactatagggaga → taatacgactcacWNNNgSRRNN), and screened for stronger mutants. |
For details, please refer to https://2019.igem.org/Team:ZJU-China/Improve. | For details, please refer to https://2019.igem.org/Team:ZJU-China/Improve. | ||
| Line 57: | Line 63: | ||
To test the function of mutant promoters, we chose eGFP as our reporter, and added a lac operator behind the promotor to control transcription starts simultaneously. When the E.coli BL21(DE3) is cultured at the stage of logarithmic phase, we added 0.5 mM IPTG to induce the expression of GFP in strains BL21(DE3) for 4 hours. By assessing the absolute fluorescence and OD600, we can conclude the relative strength of all promoters. We screened out four mutants with higher intensity. | To test the function of mutant promoters, we chose eGFP as our reporter, and added a lac operator behind the promotor to control transcription starts simultaneously. When the E.coli BL21(DE3) is cultured at the stage of logarithmic phase, we added 0.5 mM IPTG to induce the expression of GFP in strains BL21(DE3) for 4 hours. By assessing the absolute fluorescence and OD600, we can conclude the relative strength of all promoters. We screened out four mutants with higher intensity. | ||
[[File:BBa I719005 improve ZJU2019.png|center|350px|thumb|'''Figure 1. Strength of wildtype T7 promoter and mutant promoters.'''Relative fluorescent intensity is standardized with fluorescence (excitation wavelength: 485 nm; detection wavelength: 528 nm) per OD600. The intensity of the four mutants was significantly higher than that of the wild type.]] | [[File:BBa I719005 improve ZJU2019.png|center|350px|thumb|'''Figure 1. Strength of wildtype T7 promoter and mutant promoters.'''Relative fluorescent intensity is standardized with fluorescence (excitation wavelength: 485 nm; detection wavelength: 528 nm) per OD600. The intensity of the four mutants was significantly higher than that of the wild type.]] | ||
| − | As we can see from the figure, our mutant promoters showed largely increased strength compared with wild type T7 promoter. Therefore, our mutant promoters offer users more opportunity to control the expression of protein using T7 promoter and permit higher levels of target protein expression to be obtained. | + | As we can see from the figure, our mutant promoters showed largely increased strength compared with wild type T7 promoter.Therefore, our mutant promoters offer users more opportunity to control the expression of protein using T7 promoter and permit higher levels of target protein expression to be obtained. |
==III Protocol== | ==III Protocol== | ||
| Line 83: | Line 89: | ||
[1] Ikeda R A, Ligman C M, Warshamana S, et al. T7 promoter contacts essential for promoter activity in vivo[J]. Nucleic Acids Research, 1992, 20(10): 2517-2524. | [1] Ikeda R A, Ligman C M, Warshamana S, et al. T7 promoter contacts essential for promoter activity in vivo[J]. Nucleic Acids Research, 1992, 20(10): 2517-2524. | ||
[2] Paul S, Stang A, Lennartz K, Tenbusch M, Uberla K. Selection of a T7 promoter mutant with enhanced in vitro activity by a novel multi-copy bead display approach for in vitro evolution[J]. Nucleic Acids Research, 2013, 41(1):e29. | [2] Paul S, Stang A, Lennartz K, Tenbusch M, Uberla K. Selection of a T7 promoter mutant with enhanced in vitro activity by a novel multi-copy bead display approach for in vitro evolution[J]. Nucleic Acids Research, 2013, 41(1):e29. | ||
| + | |||
| + | =SUSTech_Shenzhen 2020 characterization= | ||
| + | |||
| + | Protein anti-CRISPR-associated 1 (Aca1) is expressed by T7 promoter, Aca1 will subsequently inhibit the anti-CRISPR promoter, so the gene expression downstream anti-CRISPR will be inhibited until Navitoclax (a compound that can inhibit Aca1 found by SUSTech_Shenzhen) is added artificially. Without the inhibition from Aca1, the expression of genes downstream anti-CRISPR promoter will increase significantly. Therefore, protein expression can be regulated artificially. | ||
| + | |||
| + | We characterised the efficiency of this part of promoting inhibitor in a protein regulation system, the inhibitor was sufficiently expressed to inhibite a downstream promoter, T7 promoter successfully worked in our composite part [https://parts.igem.org/Part:BBa_K3423005 BBa_K3423005]. Green fluorescent protein is used as a reporter, which can reflect the promotion effciency of Aca1 using T7 promoter: | ||
| + | https://static.igem.org/mediawiki/parts/3/33/SUSTech_ShenzhenGFP.png | ||
| + | |||
| + | =SZ-SHD 2021's characterization= | ||
| + | |||
| + | Keratinases kerAvDZ50 (<partinfo>K3895007</partinfo>), kerBteQ7 (<partinfo>K3895008</partinfo>), kerBlMKU3 (<partinfo>K3895009</partinfo>), and kerBlER-15 (<partinfo>K3895010</partinfo>) were expressed by T7 promoter. The sequences were constructed into PET 28a(+) plasmids and transformed to ''E.coli'' BL21 (DE3). | ||
| + | |||
| + | PCR was conducted first for verification of the correct plasmids. Then the amplified DNAs were sequenced (forward primer: TCGATCCCGCGAAATTAATACG; reverse primer: AGGGGTTATGCTAGTTATTGCTCA) and compared with the designed DNA sequences for further verification. | ||
| + | |||
| + | {|border=0 width="90%" align="center" | ||
| + | |-align="center" | ||
| + | |[[File:T--SZ SHD--PCR2.png|400px|thumb|center|'''Figure 1.''' PCR and gel electrophoresis of kerBlMKU3, kerBlER-15, kerAvDZ50, and kerBteQ7.The results indicated succesful transformation of these four keratinases.]] | ||
| + | |[[File:T--SZ SHD--SDSPAGE.jpg|400px|thumb|center|''' Figure 2.''' SDS-PAGE of the 4 keratinases. Lane1. Protein molecular marker; lane2. Empty PET-28a vector; lane3. Crude enzyme (kerAvDZ50); lane4. Supernatant obtained after Ni-NTA incubation; lane5. Supernatant obtained after 1st protein wash; lane6. Supernatant obtained after 2st protein wash; lane7. Empty PET-28a vector; lane8. Crude enzyme (kerBteQ7); lane9. Supernatant obtained after Ni-NTA incubation; lane10. Supernatant obtained after 1st protein wash; lane11. Empty PET-28a vector; lane12. Crude enzyme (kerBlER15); lane13. Supernatant obtained after Ni-NTA incubation; lane14. Supernatant obtained after 1st protein wash; lane15. Supernatant obtained after 2st protein wash]] | ||
| + | |} | ||
| + | |||
| + | {|border=1 width="90%" align="center" | ||
| + | |- | ||
| + | !width="20%" style="background:#CCCCFF"|Name of keratinase | ||
| + | !width="20%"|IPTG concentration/mM | ||
| + | !width="20%"|Temperature/°C | ||
| + | !width="20%"|Time/h | ||
| + | |||
| + | |-align="center" | ||
| + | |style="background:#EEEEFF"|KerBteQ7 | ||
| + | |5 | ||
| + | |16 | ||
| + | |12 | ||
| + | |-align="center" | ||
| + | |style="background:#EEEEFF"|KerAVDZ50 | ||
| + | |0.67139 | ||
| + | |16 | ||
| + | |12 | ||
| + | |-align="center" | ||
| + | |style="background:#EEEEFF"|KerBIER15 | ||
| + | |0.4196 | ||
| + | |37 | ||
| + | |16 | ||
| + | |-align="center" | ||
| + | |style="background:#EEEEFF"|KerBIMKU3 | ||
| + | |0.4 | ||
| + | |37 | ||
| + | |4 | ||
| + | |} | ||
| + | <br> | ||
| + | =SDSZ_China 2021 contribution= | ||
| + | |||
| + | T7 promoter is commonly used in iGEM projects, in our experiment, LacI was designed as a repressor and IPTG as an inducer to control the state of T7 promoter. To verify its function, we incorporated T7 promoter with LacI operon on pET28b backbone. Then the plasmid was transformed to <i>E.coli</i> top10 for test. | ||
| + | |||
| + | In the experiment, single colony was inoculated into LB medium, and cultivated vernight at 37° as seed culture. The seed culture was inoculated into LB medium as 1:100, and IPTG were added at 0h respectively. The concentration of IPTG was 0, 0.02, 0.05, 0.1, 0.2, 0.5mM, and two parallel samples for each concentration. 200 μl of each sample were taken to measure the fluorescence after 12 hours of induction. | ||
| + | |||
| + | Then, we tested OD values and the expression of GFP of two groups respectively. The graph below showed the ratio of fluorescence value/OD value of these two datas. | ||
| + | |||
| + | It is generally recognised that T7 promoter is not active in ordinary hosts, however,we found that it still maintain active functions at low concentration of IPTG (0.1mM) acordding to our results. That may be due to the potential endogenous promoter on pET28b and the gene expression caused by its high copy number. Please be cautious when using engineered strains to produce highly toxic protein expression plasmids. | ||
| + | |||
| + | [[File:T--SDSZ_China--GFP.png|700px|thumb|center]] | ||
| + | |||
| + | =Jiangnan-China 2022's contribution= | ||
| + | ==I Overview== | ||
| + | During the experiment, we tried to optimize the promoter in order to further improve the acid resistance of Escherichia coli (E. coli). We reviewed the standard components documented in the parts registry and found this T7 promoter. It is submitted by the iGEM07_Imperial team. On the basis of their study, we reviewed many relevant documents and found an article titled " Engineering an optimized expression operating unit for improved recombinant protein production in Escherichia coli ".[1] In this article, to improve recombinant protein production in E. coli, their team constructed a novel Expression Operating Unit (EOU) for recombinant protein expression in E. coli and a new optimized T7 promoter variant. Compared with the internal sequence of the wild-type T7 promoter, this new T7 promoter variant had four nucleotide mutations and the extension CCGGT was inserted in the 5′ end, while the 3' end remaining unchanged. The transcriptional activity of this new T7 promoter variant was increased (1.7-fold higher) compared to that of the typical wild-type T7 promoter sequence. At the same time, we were troubled by the inability of the natural E. coli promoter to cope with the changing and complex environment during fermentation, while the conventional inducible promoter imposed a metabolic burden on the organism and required the use of specific inducers to function. Inspired by this article, we developed the idea to modify our promoter and achieved good results in the subsequent experiments. Therefore, we share this article here, hoping to provide a reference for our friends who have not read it yet. | ||
| + | ==II Reference== | ||
| + | [1]Sara P.O. Santos, Luis Fabian S. Garcés, Filipe S.R. Silva, Leonardo F. Santiago, Carina S. Pinheiro, Neuza M. Alcantara-Neves, Luis G.C. Pacheco,Engineering an optimized expression operating unit for improved recombinant protein production in Escherichia coli,Protein Expression and Purification,Volume 199,2022,106150,ISSN 1046-5928, https://doi.org/10.1016/j.pep.2022.106150. | ||
Latest revision as of 09:01, 3 October 2022
T7 Promoter
Constitutive promoter derived from the T7 bacteriophage. Allows high expression of proteins only when the T7 polymerase is present. This part is identical to the part BBa_R0085 which currently hasn't been built.
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21COMPATIBLE WITH RFC[21]
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
Characterization in vitro
This part has been characterized for temperature sensitivity in the Cell-Free Chassis by iGEM Imperial 2007. For more detail, please check the testing [http://2007.igem.org/Imperial/Wet_Lab/Protocols/Prot1.9 protocols]and [http://2007.igem.org/Imperial/Wet_Lab/Results/Res1.9 results].

| Parameter | Value and Description |
|---|---|
| Optimum temperature | The T7 promoter has a highest output at 25°C, with a one-fold increase in GFP molecules synthesized compared to 37°C. The T7 promoter also has a minimal amount of output at 4°C. |
| Expression Life-span | The rate of GFP synthesis by the T7 promoter reaches a peak at around 30-60 minutes. |
| Peak Expression | The production of GFP decreases to minimal levels after 2 hours and tends towards nil after 4 hours. |
Team Warsaw 2010's measurement
Absolute promoter strength: 41,8pg RNA/minute/ug substrate DNA. It equals 8,92 microPoPSContribution
Group: Valencia_UPV iGEM 2018
Author: Adrián Requena Gutiérrez, Carolina Ropero
Summary: We adapted the part to be able to assemble transcriptional units with the Golden Gate assembly method
Documentation:
In order to create our complete [http://2018.igem.org/Team:Valencia_UPV/Part_Collection part collection] of parts compatible with the Golden Gate assembly method, we made the part BBa_K2656000 which is this part adapted to the Golden Gate technology.
St_Andrews 2019 characterization
We characterised this part using an in vitro T7 polymerase transcription system, measuring the concentration of Firefly Luciferase RNA transcript at 3 different temperatures (30°C, 37°C, 45°C), produced over time. Detail of the protocol can be found on our Experimentspage.
We fitted our results using a beta regression in R, with p << 0.01 for all models. Our results are plotted below. The regressed model is shown by a bold line and a 95% confidence interval is bounded by two dashed lines.
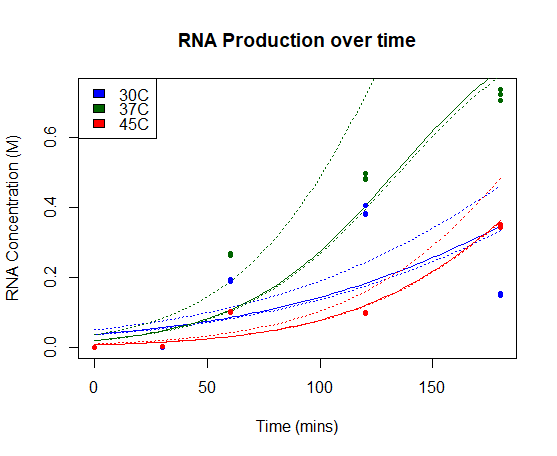
RNA Production over time: Concentration of Firefly Luciferase RNA transcript produced using T7 Polymerase and BBa_I719005 in vitro over time. Models of RNA as a function of time are shown in bold line, with 95% confidence intervals for each model bounded by two dashed lines.
Our results show a highest expression rate of transcript at 37oC when using T7 polymerase in vitro.
ZJU-China 2019's improvement
I Overview
This year team ZJU-China has upgraded four modified T7 promoters with increased strength (BBa_K2965011, BBa_K2965012, BBa_K2965013, BBa_K2965014). With the help of previous research[1-2], we carefully chose the site which would be mutated by PCR. These sites distribute in the range from -3 to +6. We mutated these sites by adding variable bases to primers (taatacgactcactatagggaga → taatacgactcacWNNNgSRRNN), and screened for stronger mutants. For details, please refer to https://2019.igem.org/Team:ZJU-China/Improve.
II Result
To test the function of mutant promoters, we chose eGFP as our reporter, and added a lac operator behind the promotor to control transcription starts simultaneously. When the E.coli BL21(DE3) is cultured at the stage of logarithmic phase, we added 0.5 mM IPTG to induce the expression of GFP in strains BL21(DE3) for 4 hours. By assessing the absolute fluorescence and OD600, we can conclude the relative strength of all promoters. We screened out four mutants with higher intensity.

As we can see from the figure, our mutant promoters showed largely increased strength compared with wild type T7 promoter.Therefore, our mutant promoters offer users more opportunity to control the expression of protein using T7 promoter and permit higher levels of target protein expression to be obtained.
III Protocol
1. Use BamHI and XhoI to insert eGFP fragment into the pET28 plasmid.
2. Linearize pET28-eGFP plasmid by PCR. (WT: F-CCCGCTGCAGTAATACGACTCACTATAGGGAGAGGAATTGTGAGCGGATAACAA / R- GCGGTGGACTGCAGCAACTCAGCTTCCTTTCGGGCT ; Mu: F-CCCGCTGCAGTAATACGACTCACWNNNGSRRNNGGAATTGTGAGCGGATAACAA / R- GCGGTGGACTGCAGCAACTCAGCTTCCTTTCGGGCT )
3. Cyclization the plasmid by PstI and ligase.
4. Transform the plasmids into E. coli BL21(DE3), add 20μl 100mM IPTG to the plate before coating the plate.
5. Incubate at 37℃ and observe colony color to screen for strong mutants preliminarily.
6. Pick greener single colonies, add the colonies into 3ml Lb, incubate at 37℃ in a shaker for 6-8h to dilute the IPTG introduced when picking single colonies.
7. Add 10μl germ solution from last step into 3ml Lb, incubate at 37℃ in a shaker until OD600 reaches 0.6.
8. Add IPTG to final concentrations 0.5mM, and induce for 4h at 18℃.
9. Measure the fluorescence (excitation wavelength: 485 nm; detection wavelength: 528 nm) and OD600.
IV Reference
[1] Ikeda R A, Ligman C M, Warshamana S, et al. T7 promoter contacts essential for promoter activity in vivo[J]. Nucleic Acids Research, 1992, 20(10): 2517-2524. [2] Paul S, Stang A, Lennartz K, Tenbusch M, Uberla K. Selection of a T7 promoter mutant with enhanced in vitro activity by a novel multi-copy bead display approach for in vitro evolution[J]. Nucleic Acids Research, 2013, 41(1):e29.
SUSTech_Shenzhen 2020 characterization
Protein anti-CRISPR-associated 1 (Aca1) is expressed by T7 promoter, Aca1 will subsequently inhibit the anti-CRISPR promoter, so the gene expression downstream anti-CRISPR will be inhibited until Navitoclax (a compound that can inhibit Aca1 found by SUSTech_Shenzhen) is added artificially. Without the inhibition from Aca1, the expression of genes downstream anti-CRISPR promoter will increase significantly. Therefore, protein expression can be regulated artificially.
We characterised the efficiency of this part of promoting inhibitor in a protein regulation system, the inhibitor was sufficiently expressed to inhibite a downstream promoter, T7 promoter successfully worked in our composite part BBa_K3423005. Green fluorescent protein is used as a reporter, which can reflect the promotion effciency of Aca1 using T7 promoter:

SZ-SHD 2021's characterization
Keratinases kerAvDZ50 (BBa_K3895007), kerBteQ7 (BBa_K3895008), kerBlMKU3 (BBa_K3895009), and kerBlER-15 (BBa_K3895010) were expressed by T7 promoter. The sequences were constructed into PET 28a(+) plasmids and transformed to E.coli BL21 (DE3).
PCR was conducted first for verification of the correct plasmids. Then the amplified DNAs were sequenced (forward primer: TCGATCCCGCGAAATTAATACG; reverse primer: AGGGGTTATGCTAGTTATTGCTCA) and compared with the designed DNA sequences for further verification.
 Figure 2. SDS-PAGE of the 4 keratinases. Lane1. Protein molecular marker; lane2. Empty PET-28a vector; lane3. Crude enzyme (kerAvDZ50); lane4. Supernatant obtained after Ni-NTA incubation; lane5. Supernatant obtained after 1st protein wash; lane6. Supernatant obtained after 2st protein wash; lane7. Empty PET-28a vector; lane8. Crude enzyme (kerBteQ7); lane9. Supernatant obtained after Ni-NTA incubation; lane10. Supernatant obtained after 1st protein wash; lane11. Empty PET-28a vector; lane12. Crude enzyme (kerBlER15); lane13. Supernatant obtained after Ni-NTA incubation; lane14. Supernatant obtained after 1st protein wash; lane15. Supernatant obtained after 2st protein wash |

| Name of keratinase | IPTG concentration/mM | Temperature/°C | Time/h |
|---|---|---|---|
| KerBteQ7 | 5 | 16 | 12 |
| KerAVDZ50 | 0.67139 | 16 | 12 |
| KerBIER15 | 0.4196 | 37 | 16 |
| KerBIMKU3 | 0.4 | 37 | 4 |
SDSZ_China 2021 contribution
T7 promoter is commonly used in iGEM projects, in our experiment, LacI was designed as a repressor and IPTG as an inducer to control the state of T7 promoter. To verify its function, we incorporated T7 promoter with LacI operon on pET28b backbone. Then the plasmid was transformed to E.coli top10 for test.
In the experiment, single colony was inoculated into LB medium, and cultivated vernight at 37° as seed culture. The seed culture was inoculated into LB medium as 1:100, and IPTG were added at 0h respectively. The concentration of IPTG was 0, 0.02, 0.05, 0.1, 0.2, 0.5mM, and two parallel samples for each concentration. 200 μl of each sample were taken to measure the fluorescence after 12 hours of induction.
Then, we tested OD values and the expression of GFP of two groups respectively. The graph below showed the ratio of fluorescence value/OD value of these two datas.
It is generally recognised that T7 promoter is not active in ordinary hosts, however,we found that it still maintain active functions at low concentration of IPTG (0.1mM) acordding to our results. That may be due to the potential endogenous promoter on pET28b and the gene expression caused by its high copy number. Please be cautious when using engineered strains to produce highly toxic protein expression plasmids.

Jiangnan-China 2022's contribution
I Overview
During the experiment, we tried to optimize the promoter in order to further improve the acid resistance of Escherichia coli (E. coli). We reviewed the standard components documented in the parts registry and found this T7 promoter. It is submitted by the iGEM07_Imperial team. On the basis of their study, we reviewed many relevant documents and found an article titled " Engineering an optimized expression operating unit for improved recombinant protein production in Escherichia coli ".[1] In this article, to improve recombinant protein production in E. coli, their team constructed a novel Expression Operating Unit (EOU) for recombinant protein expression in E. coli and a new optimized T7 promoter variant. Compared with the internal sequence of the wild-type T7 promoter, this new T7 promoter variant had four nucleotide mutations and the extension CCGGT was inserted in the 5′ end, while the 3' end remaining unchanged. The transcriptional activity of this new T7 promoter variant was increased (1.7-fold higher) compared to that of the typical wild-type T7 promoter sequence. At the same time, we were troubled by the inability of the natural E. coli promoter to cope with the changing and complex environment during fermentation, while the conventional inducible promoter imposed a metabolic burden on the organism and required the use of specific inducers to function. Inspired by this article, we developed the idea to modify our promoter and achieved good results in the subsequent experiments. Therefore, we share this article here, hoping to provide a reference for our friends who have not read it yet.
II Reference
[1]Sara P.O. Santos, Luis Fabian S. Garcés, Filipe S.R. Silva, Leonardo F. Santiago, Carina S. Pinheiro, Neuza M. Alcantara-Neves, Luis G.C. Pacheco,Engineering an optimized expression operating unit for improved recombinant protein production in Escherichia coli,Protein Expression and Purification,Volume 199,2022,106150,ISSN 1046-5928, https://doi.org/10.1016/j.pep.2022.106150.