
Difference between revisions of "Part:BBa K3187028"
JonathanFu (Talk | contribs) |
EmiliaEmily (Talk | contribs) |
||
| (27 intermediate revisions by 5 users not shown) | |||
| Line 1: | Line 1: | ||
| − | |||
__NOTOC__ | __NOTOC__ | ||
<partinfo>BBa_K3187028 short</partinfo> | <partinfo>BBa_K3187028 short</partinfo> | ||
| Line 5: | Line 4: | ||
<html> | <html> | ||
<div class="container"> | <div class="container"> | ||
| − | + | <div class="row"> | |
| − | + | <div class="col mx-2"> | |
| − | + | <h1>Profile</h1> | |
| − | + | <table style=“width:80%“> | |
| − | + | <tr> | |
| − | + | <td><b>Name</b></td> | |
| − | + | <td>Sortase A7M </td> | |
| − | + | </tr> | |
| − | + | <tr> | |
| − | + | <td><b>Base pairs</b></td> | |
| − | + | <td>450</td> | |
| − | + | </tr> | |
| − | + | <tr> | |
| − | + | <td><b>Molecular weight</b></td> | |
| − | + | <td>17.85 kDa</td> | |
| − | + | </tr> | |
| − | + | <tr> | |
| − | + | <td><b>Origin</b></td> | |
| − | + | <td><i>Staphylococcus aureus</i>, synthetic</td> | |
| − | + | </tr> | |
| − | + | <tr> | |
| − | + | <td><b>Properties</b></td> | |
| − | + | <td> Ca<sup>2+</sup>-independent, transpeptidase, linking sorting motif LPXTG to poly-glycine Tag | |
| − | + | </td> | |
| − | + | </tr> | |
| − | + | </table> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | </html> | |
| + | <!-- --> | ||
| + | <h2><span class='h3bb'>Sequence and Features</span></h2> | ||
| + | <partinfo>BBa_K3187028 SequenceAndFeatures</partinfo> | ||
| − | + | <!-- Uncomment this to enable Functional Parameter display | |
| − | + | ===Functional Parameters=== | |
| − | + | <partinfo>BBa_K3187028 parameters</partinfo> | |
| − | + | <!-- --> | |
| − | + | <html> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <h1>Structure</h1> | |
| − | + | <img class="center" src="https://2019.igem.org/wiki/images/1/18/T--TU_Darmstadt--Sortasegif4.gif" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 1 : | |
| − | + | </b> | |
| − | + | Modeled 3D-Structure of Sortase A7M. To find out more, visit our<a | |
| − | + | href="http://2019.igem.org/Team:TU_Darmstadt/Model" target="_blank"> modeling page</a>. For the PDB file of our model click | |
| − | + | <a href="https://static.igem.org/mediawiki/2019/8/88/T--TU_Darmstadt--Sortasepdb.txt" target="_blank">here</a>. | |
| − | + | </a> | |
| − | + | </p> | |
| − | < | + | </div> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | <h1>Usage and Biology</h1> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <h3>Transpeptidase: Sortase</h3> | |
| − | + | <p>Sortases belong to the class of <b>transpeptidases</b> and are mostly found in gram-positive bacteria. | |
| − | + | The high rate of resistance to several antibiotics targeting gram-positive bacteria is also based on the | |
| − | + | property of this enzyme class. Sortases can <b>non-specifically attach</b> virulence and | |
| − | + | adhesion‐associated proteins to the peptidoglycans of the cell-surface. | |
| − | + | <br> | |
| − | + | In general, sortases are divided into six groups (A-F) that have slightly different properties and | |
| − | + | perform three tasks in cells. Group A and B attach proteins to the cell-surface while Group C and D help | |
| − | + | building pilin-like structures. Group E and F are not properly investigated yet which is why their exact | |
| − | + | function is not known. | |
| − | + | For our project we are especially interested in the sortases of the <b>group A</b> since they | |
| − | + | <b>covalently attach various proteins or peptides</b> on the cell membrane as long as their targeting | |
| − | + | motif is at the C-terminus of the corresponding protein. In comparison to other transpeptidases | |
| − | + | Sortase A has the advantage that it is rather stable regarding variations in pH | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | Sortase A catalyzes the <b>formation and cleavage of a peptide bond</b> between the <b>C-terminal |
| − | + | LPXTG</b> amino acid motif and an <b>N-terminal poly-glycine</b> motif. The enzyme originates from | |
| − | + | <i>Staphylococcus aureus</i> and is able to connect any two proteins as long as they possess those | |
| − | + | matching target sequences. In the pentapeptide motif LPXTG, X can be any amino acid except cysteine. | |
| − | + | Sortase A is rather promiscuous with regard to the amino acid sequence directly upstream of | |
| − | + | this motif, a fact that makes it optimal for labeling applications. Even better, amino acids C-terminal | |
| − | + | of the poly-glycine motif are not constrained to a certain sequence. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <h3>Reaction</h3> | |
| + | <p> | ||
| + | To better understand how the enzymatic reaction works it is necessary to look at the crystal structure | ||
| + | of Sortase A. The enyzme consists of an eight-stranded β‐barrel fold structure. The <b>active | ||
| + | site</b> is hydrophobic and | ||
| + | contains the catalytic cysteine residue <b>Cys184</b> as well as a key histidine residue <b>H120</b> | ||
| + | that can form | ||
| + | a thiolate-imidazolium with the neighboring cysteine. An additional structural property that also other | ||
| + | sortases | ||
| + | show is the calcium binding site formed by the β3/β4 loop. The binding of a calcium ion slows | ||
| + | the motion | ||
| + | of the active site by coordinating to a residue in the β6/β7 loop. This helps binding the | ||
| + | substrate and | ||
| + | increasing the enzymatic activity nearly eightfold. When a substrate gets into the active site, the | ||
| + | cysteine | ||
| + | attacks the amide | ||
| + | bond between the threonine and the glycine in the <b>LPXTG motif</b>. After this the protonated | ||
| + | imidazolium serves as an | ||
| + | acid for the departing glycine with unbound NH<sub>2</sub> of the former amide bond while the rest of | ||
| + | the motif is | ||
| + | bound to the cysteine residue. Another glycine nucleophile is then necessary in its deprotonated form to | ||
| + | attack | ||
| + | the thioester and re-establish an amide bond at the LPET-motif. This reaction is dead-ended if the used | ||
| + | nucleophile | ||
| + | is water. Due to the fact | ||
| + | that the | ||
| + | mechanism is based on protonated forms of the catalytic residues the reaction is quite pH-dependent. | ||
| + | Although the | ||
| + | Sortase A in general is relatively stable between pH 3 and 11 the reaction works best around pH | ||
| + | 8. | ||
<p> | <p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | <h3>Sortase variants</h3> | ||
| + | <p>Due to the fact that the wildtype Sortase A shows rather slow kinetics, a pentamutant has | ||
| + | been | ||
| + | developed (<b>Sortase A5M</b>). | ||
| + | This version of the enzyme carries mutations in P94R/D160N/D165A/K190E/K196T which lead to a | ||
| + | 140- fold increase in activity. Thereby, reaction rates are improved even at low temperature, | ||
| + | however, Sortase A5M is still <b>Ca<sup>2+</sup>-dependent</b>. This dependence interferes | ||
| + | with | ||
| + | potential <i><b>in vivo</i> usage</b>, as the concentrations of calcium in living cells can | ||
| + | vary considerably. Hence a sortase mutant that acts across high differences in calcium | ||
| + | concentrations or even works completely <b>Ca<sup>2+</sup>-independently</b> would be required | ||
| + | for | ||
| + | <i>in vivo</i> applications of sortase. To attain a high yield enzyme which is also | ||
| + | calcium-independent Ca<sup>2+</sup>-independent mutations were combined with the | ||
| + | Sortase A5M | ||
| + | resulting in <b>Sortase A7</b> variants such as the <b>Sortase A7M</b>. The newly | ||
| + | achieved | ||
| + | calcium-independence of these variants enable sortase applications not only <i>in vitro</i> | ||
| + | but | ||
| + | <i>in vivo</i> as well. | ||
| + | </p> | ||
| + | <p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| + | <h3>Sortase A7M</h3> | ||
| + | <p>For our project we chose to work with this optimized <a | ||
| + | href="http://2019.igem.org/Team:TU_Darmstadt/Project/Sortase" | ||
| + | target="_blank"><b>Sortase A7M</b></a>. Its size is about 17.85 kDa and it has been | ||
| + | shown to be stable for several weeks in the fridge at 4 °C. It also possesses the same | ||
| + | properties of pH stability like other sortases | ||
| + | but comes with the advantage of being <b>calcium independent</b>. | ||
| + | |||
| + | "Sortagging" applications have included the cyclization of proteins and peptides | ||
| + | , modification and labeling of antibodies and the synthesis of protein conjugates with | ||
| + | drugs, peptides, peptide nucleic acids and sugars.Moreover it poses a lot of advantages for | ||
| + | the <b>binding of two proteins | ||
| + | <i>in vivo</i></b> since it has relatively small tags which avoids putting too much | ||
| + | metabolic burden on the cells when expressing the proteins of interest. This also avoids | ||
| + | disturbing the folding of the proteins of interest and the later biological functions since | ||
| + | the | ||
| + | Sortase A7M is able to work under <b>physiological conditions</b>. Other methods like | ||
| + | the intein- based labeling of surfaces require large fusion-proteins | ||
| + | with the intein domain which puts stress on the living cells and might cause folding and | ||
| + | solubility issues. Another application for sortase-mediated systems is the anchoring of | ||
| + | proteins | ||
| + | on the cell wall of gram-positive bacteria which can be used for display of heterologous | ||
| + | proteins. It is also possible to attach non-biological molecules to the respective tag. The | ||
| + | accessibility and flexibility determine the ability of a sortase enzyme to recognize the | ||
| + | sorting | ||
| + | motif and catalyzing the transacylation.</p> | ||
| − | + | <h1>Methods</h1> | |
| − | + | <h3>Cloning</h3> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | |
| − | + | ||
| − | + | ||
| − | < | + | |
| − | + | ||
| − | + | ||
| + | <p>The methods used for cloning of the different mutants of the sortase were restriction and | ||
| + | ligation via <i>NdeI</i> and <i>SalI</i> and Gibson assembly. The vector posesses a | ||
| + | kanamycin resistance and the <i>srta7m</i> is controlled | ||
| + | through a T7 promoter, | ||
| + | which can be induced with IPTG. Sortase A7M is controlled by the same T7 promoter. | ||
| + | The product was checked via sequencing. | ||
| + | </p> | ||
| − | + | <h3>Expression and purification</h3> | |
| − | + | <p> | |
| − | + | After successfully transforming our sortase genes in BL21 cells, we inoculated 100 mL | |
| − | + | overnight cultures, with the respective antibiotic. The next day 1 L cultures were | |
| − | + | inoculated with the overnight culture to reach OD<sub>600</sub> = 0.1. | |
| − | + | Subsequently | |
| − | + | the cultures were incubated under constant shaking at 37 °C until they reached | |
| − | + | OD<sub>600</sub> = 0.6. At OD<sub>600</sub> = 0.6 the cultures were | |
| − | + | induced | |
| + | with 0.5 mL of 1 M Isopropyl-β-D-thiogalactopyranosid (IPTG). The gene expression | ||
| + | was | ||
| + | performed at 30 °C under constant shaking overnight. After expression of | ||
| + | Sortase A7M in BL21 cultures the | ||
| + | cells | ||
| + | were crushed via EmulsiFlex (Avestin) and proteins were purified through affinity | ||
| + | chromatography | ||
| + | via Fast Protein Liquid Chromatography (FPLC) with the ÄKTA pure (GE Healthcare, Illinois, | ||
| + | USA). | ||
| + | His-Tag was used for purification of Sortase A7M and Sortase A (Stockholm) and | ||
| + | Strep-Tag II was used for purification of Sortase A5M. | ||
| + | </p> | ||
| − | + | <h3>SDS-Page</h3> | |
| − | + | <p>To verify the successful production of of Sortase A7M and others | |
| − | + | SDS-PAGEs were performed. The resulting bands were compared to the molecular weight of the | |
| − | + | different sortase variants. | |
| − | + | Also, SDS-PAGEs were completed to verify enzymatic activity in assays prior to measuring | |
| − | + | sortase | |
| − | + | properties via Fluorescence Resonance Energy Transfer (FRET). | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | <h3>Flourescence Resonance Energy Transfer (FRET)</h3> | |
| − | + | <p> | |
| − | + | To determine the kinetics of our transpeptidase variants, FRET assays were performed in 384 | |
| − | + | well-plates (dark) using a Tecan plate reader. A FRET relies on the phenomenon that an | |
| − | + | excited | |
| − | + | fluorophore (donor) transfers energy to another fluorophore (acceptor), thereby exciting it. | |
| − | + | This process only works if both fluorescent molecules are in close proximity and depends on | |
| − | + | the | |
| − | + | FRET-Pair. By transferring the energy from donor to acceptor, the donor's emission is | |
| − | + | reduced | |
| − | + | and the intensity of the acceptors emission is increased . The efficiency depends on the | |
| − | + | distance between the fluorophore, the orientation and the | |
| − | + | spectral characteristics . You can see the principle of FRET in <b>Fig. 2</b>. | |
| − | + | </p> | |
| − | + | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/8/8b/T--TU_Darmstadt--FRET.jpeg" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 2 : | |
| − | < | + | </b> |
| − | + | Jablonski diagram showing the energy transfer between a FRET-pair (inspired by | |
| − | </ | + | <a href="http://en.wikipedia.org/wiki/F%C3%B6rster_resonance_energy_transfer#/media/File:FRET_Jablonski_diagram.svg" |
| − | + | target="_blank">Alex M. Mooney</a>). | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | ||
| − | + | <h3>Mass Spectrometry</h3> | |
| − | + | <p>To estimate the product yield of catalyzed reactions by Sortase A7M we performed mass | |
| − | + | spectrometry. The tested molecules can be distinguished between products and educts due to | |
| − | + | desorption and ionization. Therefore, we used the electrospray ionization (ESI) technique | |
| − | + | for | |
| − | < | + | the mass spectrometry. This technique has a low resolution but is a very soft ionization |
| − | + | method, | |
| − | + | which makes it an optimal method for biological molecules.</p> | |
| − | + | ||
| − | + | <h3 id="ELISA">Enzyme-linked Immunosorbent Assay (ELISA)</h3> | |
| − | + | <p> | |
| − | + | The enzyme-linked immunosorbent assay (ELISA) is an analytical assay frequently utilized for | |
| − | + | immobilization and verification of different macro molecules. Immobilizing the recognition | |
| − | + | tag of the sortase on a surface allows us to verify the coupling efficiency of | |
| − | + | Sortase A7M under certain conditions. | |
| − | + | Firstly, we functionalized paper to presenting a poly-G peptide sequence on the surface. | |
| + | Using | ||
| + | the Sortase A7M, a ZZ-domain carrying a LPETG amino acid sequence is coupled to the | ||
| + | short peptide | ||
| + | sequence of GGGßA. The ZZ-domain itself shows high affinity to the human IgG antibody | ||
| + | FC-domain and | ||
| + | therefore allows the following immobilization of IgG. The secondary antibody is an | ||
| + | anti-human IgG, Fab-specific antibody carrying the horseradish | ||
| + | peroxidase enzyme (HRP). HRP is capable of converting 4-chloro-1-naphthol to | ||
| + | benzo-4-chlorocyclohexandienone | ||
| + | using hydrogen peroxide. This color-reaction allows us to draw a conclusion about the | ||
| + | previous ZZ-domain’s | ||
| + | sortase-mediated coupling efficiency since the turnover of the HRP is directly connected to | ||
| + | the ligated ZZ-domains. | ||
| + | </p> | ||
| + | |||
| + | <h1>Results</h1> | ||
| + | <h2>Characterization of Sortase A7M (and comparison to <a | ||
| + | href="https://parts.igem.org/Part:BBa_K3187016" target="_blank">BBa_K3187016</a>) | ||
| + | </h2> | ||
| + | <h3>How do we measure if our purified sortases are active?</h3> | ||
| + | <p>After purification of the sortases, we first performed SDS-PAGEs to verify that they are pure | ||
and | and | ||
| − | + | monomeric. You can see in <b>Fig. 3</b> that the purifications were successful. Next, we | |
| − | + | tested | |
| − | and | + | if the purified sortases connect two proteins that carry the important Sortase-recognition |
| − | + | tags, | |
| − | + | N-terminal polyG and C-terminal LPETGG. Therefore, we added the sortases to a mix of | |
| − | + | GGGG-mCherry and mCherry-LPETGG. The reactions were performed in different buffers, at | |
| − | + | different | |
| + | enzyme-to-substrate ratios and for different time spans. We performed an SDS-PAGE, and prior | ||
| + | to | ||
| + | Coomassie staining, we recorded fluorescent images of the gel. Thereby, we could identify | ||
| + | mCherry bands in the gel. | ||
| + | </p> | ||
| + | <img class="center" | ||
| + | src="https://2019.igem.org/wiki/images/5/59/T--TU_Darmstadt--SDS_Sortase_A7M_Sortase_A5M.png" | ||
| + | style="max-width:30%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 3 : | ||
| + | </b> | ||
| + | SDS-PAGE of Sortase A7M and Sortase A5M where the bands show up at | ||
| + | approximately 15 kDa. Our estimated size for Sortase A7M was 17.85 kDa, | ||
| + | and for Sortase A5M 18.07 kDa. This confirms the result shown on the gel, | ||
| + | since the band of Sortase A5M is a little higher than the one of Sortase A7M. | ||
| + | </p> | ||
| + | </div> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/6/67/T--TU_Darmstadt--SDS_buff_fluor_Srt5_7_1%3B3_1%3B10_.png" | ||
| + | style="max-width:50%" /> | ||
| + | </a> | ||
| − | </ | + | <img class="img-fluid center" |
| − | caption | + | src="https://2019.igem.org/wiki/images/0/0d/T--TU_Darmstadt--SDS_buff_both_Srt5_7_1%3B3_1%3B10_.png" |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 4 : | |
| + | </b> | ||
| + | <b>a)</b> Fluorescence gel of the sortase-reaction of GGGG-mCherry and mCherry-LPETGG | ||
| + | mediated by Sortase A7M incubated for 2 h and | ||
| + | 4 h each. Reaction solutions were mixed with different ratios from enzyme to | ||
| + | substrate concentration(1:3;1:10) and each incubated in two different buffers(Tris-HCl | ||
| + | and Ammoniumdicarbonat). | ||
| + | Product bands at a height of about 57 kDa can be seen in lane 4, 5, 6, 8, 9 (from | ||
| + | left to right). The bands below the product at about 38 kDa could be semi-denatured | ||
| + | mCherry dimers.<br> | ||
| + | <b>b)</b> Fluorescence gel on top of the coomassie-stained gel of the sortase-reaction | ||
| + | of GGGG-mCherry and mCherry-LPETGG mediated by Sortase A7M incubated for 2 h | ||
| + | and | ||
| + | 4 h each. Reaction solutions were mixed with different ratios from enzyme to | ||
| + | substrate concentration(1:3;1:10) and each incubated in two different buffers (Tris-HCl | ||
| + | and Ammoniumdicarbonat). | ||
| + | Product bands at a height of about 57 kDa can be seen in lane 4, 5, 6, 8, 9 (from | ||
| + | left to right). The bands below the product at about 38 kDa could be semi-denatured | ||
| + | mCherry dimers. Additionally, Sortase A7M can be seen at 17 kDa.The | ||
| + | unprocessed mCherry monomers can be seen at 28 kDa. | ||
| + | </p> | ||
| + | <p> | ||
| + | As shown in <b>Fig. 4</b>, under certain conditions, a product band appeared at the | ||
| + | expected | ||
| + | size of 57.3 kDa (28.5+28.8 kDa). From this first activity test, we draw three | ||
| + | conclusions: | ||
| + | </p> | ||
| − | + | <ul> | |
| − | + | <li> | |
| − | + | <b>Our purified Sortase A7M is active</b> | |
| − | + | </li> | |
| − | + | <li> | |
| − | + | <b>The enzyme-substrate ratio affects the product yield</b> | |
| − | + | </li> | |
| − | + | <li> | |
| − | + | <b>The duration of the reaction affects the product yield</b> | |
| − | + | </li> | |
| − | + | </ul> | |
| − | + | <p> | |
| − | + | <br> | |
| − | + | Additionally, TRIS buffer seems to alter the coomassie staining efficiency of Sortase | |
| − | + | A7M. | |
| − | + | This endpoint measurement gave us a first impression that our Sortase A7M works nicely. | |
| − | + | Of | |
| + | course, we wanted to further characterize the parameters of the reaction. When we | ||
| + | understand | ||
| + | the Sortase better, modification of our VLPs will become more straightforward. | ||
| − | + | </p> | |
| − | + | </div> | |
| − | + | <h3>How do we measure sortase reaction kinetics?</h3> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<p> | <p> | ||
| − | + | In the above described assays, we noticed the impact of enzyme-substrate ratio and reaction | |
| − | + | duration on the | |
| − | + | overall product yield. We thought about how to further measure the kinetics of the sortase | |
| − | + | reaction. In the | |
| − | + | literature, sortase reaction kinetics are often measured by FRET-assays. Therefore, we | |
| − | + | designed | |
| − | + | a suitable | |
| − | + | FRET-assay. | |
| − | + | ||
</p> | </p> | ||
| − | + | <h4>Development of a new FRET pair</h4> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<p> | <p> | ||
| − | + | For characterization of the reaction kinetics of Sortase A7M, Sortase A5M and | |
| − | + | Sortase A, we | |
| − | + | decided to develop a suitable FRET pair. | |
| − | + | In order to find an optimal FRET pair, we first recorded an emission and absorption | |
| − | + | spectrum of | |
| + | 5-Carboxytetramethylrhodamin-LPETG (TAMRA) and GGGG-mCherry to verify the suitability for | ||
| + | the | ||
| + | FRET effect, checking for a possible overlap between the donor's emission and the | ||
| + | acceptor's | ||
| + | excitation. | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/d/d4/T--TU_Darmstadt--TAMRA_mCherry.JPG" | |
| − | + | style="max-width:80%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 5 : | |
| − | + | </b> | |
| − | + | Design of a FRET-pair of 5-TAMRA-LPETG (TAMRA) and GGGG-mCherry (mCherry). In | |
| − | + | this configuration TAMRA acts as donor and mCherry as acceptor. When the two | |
| − | + | fluorophores are not linked via the substrates of the sortase only TAMRA is being | |
| − | + | excited. After sortase mediated ligation of the two substrates mCherry is the | |
| − | + | fluorophore being excited via the FRET and the emission of mCherry intensifies. | |
| − | + | Meanwhile, the emission of TAMRA decreases. | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<p> | <p> | ||
| − | + | TAMRA is a chemical fluorophore that has an absorbance maximum at 543 nm and an | |
| − | + | emission | |
| − | + | maximum at | |
| − | + | 570 nm. The | |
| − | + | terminal carboxy | |
| − | + | group of the dye was linked via a lysine linker to the LPETG sequence (<b>see | |
| − | + | Fig. 5</b>). | |
| − | + | mCherry has | |
| − | + | an N-terminal poly-glycine sequence and can therefore be linked to the LPETG motif of TAMRA | |
| − | + | via | |
| − | < | + | the |
| − | + | Sortase A. For a sufficient FRET-effect, it is also necessary that the distance between | |
| + | donor and | ||
| + | acceptor is lower than the Förster radius. | ||
| + | <br> | ||
| + | First, we wanted to identify which concentrations are needed for our experiment, then set up | ||
| + | the | ||
| + | reaction | ||
| + | and measured fluorescence intensities. Over time, a decline in the emission of TAMRA can be | ||
| + | observed as | ||
| + | Sortase A7M/A5M is converting more educts to products. | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/0/06/T--TU_Darmstadt--mCherry_TAMRA_Extinction_Emission.png" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 6 : | |
| − | + | </b> | |
| − | + | The graph shows the excitation and emission spectra of TAMRA and mCherry. Due | |
| − | + | to the large overlap of TAMRA emission and mCherry excitation it is possible to perform | |
| − | + | a FRET with this pair of fluorophores. The graph show the relative fluorescence unit | |
| − | + | (RFU[%]) in relation to the excited/emitted wavelength [nm]. The peaks are normalized | |
| − | + | to 100 %. | |
| − | + | </p> | |
| + | </div> | ||
<p> | <p> | ||
| − | + | The emission and excitation spectra of TAMRA and mCherry exhibit an overlap of emission of | |
| − | + | TAMRA | |
| − | + | and | |
| − | + | excitation of mCherry. Based on this output, a FRET-assay for the kinetics of | |
| + | Sortase A7M | ||
| + | was performed | ||
| + | to confirm whether the FRET-pair is working. | ||
| + | As TAMRA is excited with light of a lower wavelength than mCherry, the former serves as FRET | ||
| + | donor and the | ||
| + | latter as acceptor. We chose the excitation wavelength at 485 nm to prevent unnecessary | ||
| + | “leak” | ||
| + | excitation of mCherry. | ||
| + | Nevertheless, an excitation of mCherry could not be excluded and may have negative effects | ||
| + | on | ||
| + | the visibility | ||
| + | of the FRET. | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/b/b1/T--TU_Darmstadt--mCherry_TAMRA_Bleaching_Negativecontrol.png" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 7 : | |
| − | + | </b> | |
| − | + | Spectrum of the negative control of TAMRA and mCherry, | |
| − | + | without Sortase A7M, over the course of 20 min in 5 min intervals. | |
| − | + | Depicted are the emission wavelengths against the RFU. | |
| − | + | </p> | |
| − | </ | + | </div> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/f/fd/T--TU_Darmstadt--mCherry_TAMRA_Bleaching_Positive.png" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 8 : | |
| − | + | </b> | |
| − | + | Spectrum of TAMRA and mCherry, with Sortase A7M, over the course of | |
| − | + | 20 min in 5 min intervals. Depicted are the emission wavelengths against the | |
| − | + | RFU. The sortase-mediated ligation results in a decline of both emission peaks. | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
<p> | <p> | ||
| − | <b> | + | The analysis of the data shown in <b>Fig. 7</b> confirmed the aforementioned |
| − | + | suspicion that mCherry is also excited at 485 nm, which makes differentiation | |
| − | + | of the fluorescence more difficult. Furthermore, <b>Fig. 8</b> shows that the | |
| − | + | difference in the decline of TAMRA is not significant. Accordingly, a decline in the | |
| − | + | emission maximum of TAMRA over time is also visible in the negative control. One | |
| − | + | reason might be bleaching of TAMRA through the excitation by the laser. | |
| − | + | Nevertheless, conversion by the Sortase A7M can be observed by comparing the | |
| + | results with the negative control. | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/9/92/T--TU_Darmstadt--mCherry_TAMRA_FRET_SortaseA7M.png" | |
| − | + | style="max-width:60%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 9 : | |
| − | + | </b> | |
| − | + | Sortase reaction in TAMRA-mCherry-FRET after subtracting the negative control. Depicted | |
| − | + | is the difference in RFU over time [min]. Within the first 20 min of the substrate | |
| − | + | conversion is the quickest. At 30 min a plateau is reached. After 60 min | |
| − | + | starts catalyzing the reverse reaction. The mean ΔRFU value was normalized to zero for | |
| − | + | better visualization. The ΔRFU refers to the difference between the negative control | |
| − | + | without the respective sortase at 570 nm. | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | ||
<p> | <p> | ||
| − | + | To confirm the functionality of the Sortase A7M, another more sufficient FRET-pair was | |
| − | + | developed. The measured absorbance and emission spectra indicated that TAMRA and superfolder | |
| − | + | green fluorescence protein (sfGFP) are a possible FRET-pair. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/9/90/T--TU_Darmstadt--FRET_pair_sfGFP_TAMRA.png" | |
| − | + | style="max-width:60%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 10 : | |
| − | + | </b> | |
| − | + | The graph shows the excitation and emission spectra of | |
| − | + | TAMRA and mCherry. Due to the large overlap of sfGFP emission and TAMRA | |
| − | + | excitation it is possible to perform a FRET with this pair of fluorophores. | |
| − | + | The graph show the relative fluorescence unit (RFU[%]) in relation to the | |
| − | + | excited/emitted | |
| − | + | wavelength [nm]. The peaks are normalized to 100 %. | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/c/c0/T--TU_Darmstadt--Tamra_GFP_Johny.jpg" | |
| − | + | style="max-width:80%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 11 : | |
| − | + | </b> | |
| − | + | Design of a FRET-pair of 5-TAMRA-LPETG (TAMRA) and GGGG-sfGFP (sfGFP). In this | |
| + | configuration sfGFP acts as donor and TAMRA as acceptor. When the two fluorophores are | ||
| + | not linked only sfGFP is being excited. After sortase-mediated ligation of the two | ||
| + | substrates, TAMRA is the fluorophore being excited via FRET and the emission of TAMRA | ||
| + | intensifies. Meanwhile, the emission of sfGFP decreases. | ||
| + | </p> | ||
| + | </div> | ||
<p> | <p> | ||
| − | + | The transfer of energy from sfGFP to TAMRA can be seen by the decrease in emission of sfGFP | |
| − | + | and | |
| − | + | increase in emission from TAMRA. Compared to TAMRA as an acceptor, the sfGFP bleaches | |
| − | + | significantly less and is consequently more suitable as a donor for FRET. Furthermore, the | |
| − | + | afore | |
| + | mentioned problem of simultaneous donor and acceptor excitation seems to be solved. It seems | ||
| + | that we have found a FRET-pair with superior properties. | ||
</p> | </p> | ||
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/8/85/T--TU_Darmstadt--Sortase_A7M_Proof_N2_Without_Sortase_A7M.png" | |
| − | + | style="max-width:50%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 12 : | |
| − | + | </b> | |
| + | Spectrum of the negative control of TAMRA and sfGFP, | ||
| + | without Sortase A7M, over the course of 25 min in 5 min intervals. | ||
| + | Depicted are the emission wavelengths against the RFU. | ||
| + | </p> | ||
| + | </div> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/0/04/T--TU_Darmstadt--Sortase_A7M_Proof_N2_With_Sortase_A7M.png" | ||
| + | style="max-width:50%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 13 : | ||
| + | </b> | ||
| + | Spectrum of TAMRA and sfGFP, with Sortase A7M, over the course of | ||
| + | 25 min in 5 min intervals. Depicted are the emission wavelengths against the | ||
| + | RFU. The sortase-mediated ligation results in a decline of both emission peaks. | ||
| + | </p> | ||
| + | </div> | ||
<p> | <p> | ||
| − | + | Due to the collected data of both FRET-pairs we decided to use the TAMRA-LPETG and | |
| − | + | GGGG-sfGFP | |
| − | + | FRET-pair for further characterization of our Sortase A variants. Two reasons justify | |
| − | + | this | |
| − | + | decision: | |
</p> | </p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
<p> | <p> | ||
| − | < | + | <ul> |
| − | + | <li>TAMRA bleaches stronger than sfGFP when excited with a laser.</li> | |
| − | + | <li>The spectral overlap between TAMRA and mCherry disturbs “clean” energy transfer, | |
| − | used to | + | thus |
| − | + | the FRET-effect would be less visible and could not be used for analysis of the | |
| − | + | sortase-mediated reaction. </li> | |
| − | + | </ul> | |
| − | + | <p> | |
| − | + | For recording of sortase reaction parameters we recommend using the FRET-pair | |
| − | + | sfGFP-TAMRA. | |
| − | + | As this pair of fluorophores proved to have near perfectly aligned spectra and since the | |
| + | bleaching effect is visibly lower on sfGFP than on TAMRA, we chose to use this FRET-pair | ||
| + | in | ||
| + | most of our following assay. Nevertheless, we do not rule out the use of TAMRA-mCherry | ||
| + | as a | ||
| + | FRET-pair since we used it in several FRET-assays as well. | ||
| + | </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/a/a3/T--TU_Darmstadt--FRET_mCherry_TAMRA_Gif.png" | ||
| + | style="max-width:50%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 14 : | ||
| + | </b> | ||
| + | Animation of Sortase A7M enzyme kinetics over the course of 3 h. The | ||
| + | reaction speed increases radically in the beginning moving from RFU 8000 to RFU 6000 | ||
| + | at λ = 550 nm where a plateau is reached (blue). The negative control | ||
| + | (orange) is also reduced in its RFU due to bleaching. Nevertheless, a peak at | ||
| + | λ = 580 nm arises already after short reaction time. This peak | ||
| + | indicates the successful Fluorescence Resonance Energy Transfer. | ||
| + | </p> | ||
| + | </div> | ||
| + | <h3>Why are enzyme-substrate ratio and duration important parameters of the sortase | ||
| + | reaction?</h3> | ||
| + | <p> | ||
| + | In one of our first FRET experiments, we addressed the simple theory: More sortase in | ||
| + | the | ||
| + | reaction mix improves the initial product formation. For this, we used the | ||
| + | TAMRA-LPETG : GGGG-mCherry FRET pair. We measured the FRET change over time in | ||
| + | a | ||
| + | multiwell platereader <b>(Fig. 15)</b>. | ||
| + | </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/0/03/T--TU_Darmstadt--mCherry_TAMRA_FRET_different_concentrations.png" | ||
| + | style="max-width:50%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 15 : | ||
| + | </b> | ||
| + | Reaction kinetics of Sortase A7M in different concentrations at same level of | ||
| + | substrate | ||
| + | concentration (6 µM TAMRA-LPETG and (M)GGGG-mCherry). The light blue graph shows the | ||
| + | reaction when the | ||
| + | sortase concentration is higher (5 µM) and lower concentration (1 µM), in the dark | ||
| + | blue graph. Wen the | ||
| + | enzyme concentration is lower the maximum in substrate conversion is reached later. | ||
| + | The light blue graph | ||
| + | also shows a slight decline in product concentration from 60 min onwards.The | ||
| + | ΔRFU refers to the | ||
| + | respective negative control without each sortase at 570 nm. The mean ΔRFU value was | ||
| + | normalized to zero | ||
| + | for better visualization. | ||
| + | </p> | ||
| + | </div> | ||
| + | <p> | ||
| + | However, in this assay we observed a striking feature of the sortase reaction. In the | ||
| + | reaction with more Sortase A7M present, the FRET change started to decrease after a | ||
| + | certain | ||
| + | maximum was reached! We suspected some kind of dead-end product formation, as the | ||
| + | sortase | ||
| + | does also catalyze the reverse reaction of product to educts. Therefore, the overall | ||
| + | reaction duration is a very important parameter. We gathered more details about the role | ||
| + | of | ||
| + | the reverse reaction during our comparison of Sortase A7M and Sortase A5M. Just keep | ||
| + | reading | ||
| + | if you want to know more! | ||
| + | </p> | ||
| + | <h3>Who wins - Sortase A7M or Sortase A5M</h3> | ||
| + | <p> | ||
| + | In our introduction we described that Sortase A7M and Sortase A5M are both | ||
| + | fascinating enzymes, | ||
| + | although each of them has a unique „selling point“. Sortase A5M is faster, whereas | ||
| + | Sortase A7M | ||
| + | is Ca<sup>2+</sup>-independent. We confirmed both of these points in extensive | ||
| + | FRET-assays. | ||
| + | According to the literature, Sortase A5M works best with a | ||
| + | Ca<sup>2+</sup>-concentration of 2 mM. | ||
| + | In contrast, Sortase A7M is a calcium-independent mutant of the enzyme. Moreover, | ||
| + | Ca<sup>2+</sup> even seems to inhibit this enzyme variant slightly | ||
| + | . | ||
| + | </p> | ||
| + | <p> Firstly, we confirmed that in contrast to Sortase A5M, <b>Sortase A7M is | ||
| + | Ca<sup>2+</sup>-independent.</b> | ||
| + | The results are shown in <b>Fig. 16</b> | ||
| + | Sortase A7M also works in presence of Ca<sup>2+</sup>, but these FRET experiments | ||
| + | made | ||
| + | us | ||
| + | suspect that Ca<sup>2+</sup> may even inhibit Sortase A7M. | ||
| + | </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/9/97/T--TU_Darmstadt--FRET_SrtA7M_mitCa2%2B.png" | ||
| + | style="max-width:60%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 16 : | ||
| + | </b> | ||
| + | Sortase A7M FRET-assay of connecting TAMRA-LPETG with GGGG-sfGFP with and | ||
| + | without | ||
| + | Ca<sup>2+</sup>. The | ||
| + | Sortase A7M reaction was measured with 6 mM Ca<sup>2+</sup> every minute. | ||
| + | Sortase A7M reaction without Ca<sup>2+</sup> was measured every three | ||
| + | minutes. It is shown that this enzyme variant works with calcium and without calcium | ||
| + | as well, | ||
| + | although it seems | ||
| + | like Sortase A7M is slightly inhibited due to the presence of calcium which | ||
| + | explains why | ||
| + | the left graph is lower | ||
| + | than the right one. The ΔRFU refers to the respective negative control without each | ||
| + | sortase at | ||
| + | 514 nm. The mean ΔRFU value of the duplicates was normalized to zero for better | ||
| + | visualization. | ||
| + | </p> | ||
| + | </div> | ||
| − | + | </div> | |
| − | <p> | + | <p> |
| − | + | According to the results of this assay, Sortase A7M is definitely | |
| − | </p> | + | Ca<sup>2+</sup>-independent, since it shows |
| − | + | linking activity without calcium in the vicinity. The enzyme mutant also works in | |
| − | <p> | + | presence |
| − | + | of Ca<sup>2+</sup>, | |
| − | </p> | + | but these FRET experiments made us suspect that Ca<sup>2+</sup> may even inhibit |
| − | + | Sortase A7M, since it shows less activity with calcium around than without calcium. | |
| − | <img class="img-fluid center" | + | </p> |
| − | + | <p> | |
| + | To better address this question, an ELISA was | ||
| + | performed. Therefore, a piece of paper functionalized with GGGβA was connected to a | ||
| + | protein | ||
| + | domain, which binds | ||
| + | antibodies to the LPTEG-tag. The results are shown in <b>Fig. 18</b>. | ||
| + | </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/7/77/T--TU_Darmstadt--Absorbance_450_Sortase_yield.png" | ||
| + | style="max-width:30%" /> | ||
</a> | </a> | ||
<div class="caption"> | <div class="caption"> | ||
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 18 : | |
| − | + | </b> | |
| − | + | Absorbance at 450 nm at a temperature of 23.8˚ C <br> | |
| + | In well 1 additional 10 mM Ca<sup>2+</sup> were | ||
| + | added which was not the case in well 2. Well 3 serves as a negative control since | ||
| + | the enzyme is missing in this reaction | ||
</p> | </p> | ||
</div> | </div> | ||
| − | + | <p> | |
| − | <img class="img-fluid center" | + | As shown in <b>Fig. 18</b>, the highest absorption was measured in well 2. Thus, |
| − | + | Sortase A7M works more efficiently | |
| + | when no Ca<sup>2+</sup> is around. The absorption is also relatively high for the | ||
| + | negative | ||
| + | control, | ||
| + | which can be explained by poor washing before the substrate for Horeseradish peroxidase | ||
| + | (HPR) was added. This assay shows | ||
| + | the functionality of Sortase A7M even in context of surfaces since we confirmed | ||
| + | that | ||
| + | Sortase A7M | ||
| + | is able to connect tags attached to paper. This shows that the surface structure is not | ||
| + | a | ||
| + | relevant factor for the enzyme. | ||
| + | </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/c/c7/T--TU_Darmstadt--Vergleich_A7M_A5M_bei_opt_Bedingung.png" | ||
| + | style="max-width:60%" /> | ||
</a> | </a> | ||
<div class="caption"> | <div class="caption"> | ||
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 19 : | |
| − | + | </b> | |
| − | + | Comparison of the reaction speed of Sortase A5M with Ca<sup>2+</sup> and | |
| + | Sortase A7M without Ca<sup>2+</sup>, therefore each working under optimal conditions. | ||
| + | The kinetics were measured via a FRET connecting TAMRA-LPETG and GGGG-sfGFP. The ΔRFU refers | ||
| + | to the respective negative control without each sortase at 514 nm. The mean ΔRFU value of | ||
| + | the triplicates was normalized to zero for better visualization. | ||
</p> | </p> | ||
</div> | </div> | ||
| − | + | <p> | |
| − | <p> | + | When we compare the reaction speed of Sortase A5M and Sortase A7M, |
| − | + | Sortase A5M is the | |
| − | + | clear winner (see <b>Fig. : 19</b>). However, this means of course that the reverse | |
| − | + | reaction is also faster in the | |
| − | + | case of Sortase A5M. | |
| − | + | Consequently, Sortase A7M is the best variant for in vivo modification of our VLPs | |
| − | + | as | |
| − | < | + | it is Ca<sup>2+</sup>-independent. |
| − | + | On the other hand, Sortase A5M is a suitable | |
| − | < | + | enzyme variant for in vitro modification due to its high efficiency. |
| − | + | </p> | |
| − | < | + | <h3>What about other substrates?</h3> |
| − | + | <h4>Primary Amines</h4> | |
| − | + | <p>The literature | |
| − | </ | + | describes Sortase A7M as somewhat „promiscuous“ towards other substrates than |
| − | <p> | + | GGGG(polyG) as long as the substrate possesses a primary amine. The |
| − | + | Sortase A7M used for this assay was stored in the fridge at 4 °C for two | |
| − | + | weeks. | |
| − | + | The substrates were TAMRA with a KLPETG bound to TAMRA via the lysine side chain and | |
| − | + | 3-azidopropanamine as the example for a primary amine. The reaction was performed for | |
| − | + | two | |
| − | + | hours at 37 °C. <br> | |
| − | + | It was then analyzed by electron spray ionization mass spectrometry | |
| − | + | (ESI-MS) <b>(Fig. 20)</b>. </p> | |
| − | + | <img class="img-fluid center" | |
| − | + | src="https://2019.igem.org/wiki/images/9/96/T--TU_Darmstadt--SortaseFigure1TAMRA_KE.png" | |
| − | + | style="max-width:100%" /> | |
| − | + | ||
| − | < | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | + | ||
| − | <img class="img-fluid center" | + | |
| − | + | ||
</a> | </a> | ||
<div class="caption"> | <div class="caption"> | ||
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 20 : | |
| − | + | </b> | |
| − | + | Mass spectrum before the reaction of TAMRA-LPETG with 3-azidopropanamine | |
| + | showing the educt at 1054 g/mol. | ||
</p> | </p> | ||
</div> | </div> | ||
| − | + | <p><b>Fig. 20</b> shows the educt-peak in the mass spectrum. TAMRA with the LPETG-tag | |
| − | + | weighs 1054 g/mol. Shown above in green is the single charged molecule at 1054.27 g/mol | |
| − | <p> | + | and |
| − | + | the double charged molecule at 528.75 g/mol.</p> | |
| − | < | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/7/7a/T--TU_Darmstadt--SortaseFigure2TAMRA_KE.png" | |
| − | + | style="max-width:90%" /> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | + | ||
| − | <img class="img-fluid center" | + | |
| − | + | ||
</a> | </a> | ||
<div class="caption"> | <div class="caption"> | ||
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 21 : | |
| − | + | </b> | |
| − | + | Mass spectrum after the reaction of TAMRA-LPETG with 3-azidopropanamine | |
| + | showing the product at 1079.37g/mol. | ||
</p> | </p> | ||
</div> | </div> | ||
| − | + | <b>Fig. 21</b> shows the product-peak in the mass spectrum. The primary amine that was taken | |
| − | + | as an example has a molecular weight of 100 g/mol. After the reaction the glycine of the | |
| − | <img class="img-fluid center" | + | LPETG-tag has been removed and therefore the product only consists of |
| − | + | TAMRA-KLPET-3-azidopropanamine. When adding the two molecular weights and subtracting the | |
| + | weight | ||
| + | of the glycine it adds up to a total weight of 1078 g/mol which can be seen in the single | ||
| + | loaded | ||
| + | 1079.37 g/mol peak <b>(Fig. 21)</b>, since the ESI-MS we used has a small error margin. The | ||
| + | peak | ||
| + | in black again is the double loaded peak at 541.55 g/mol. This clearly shows that the | ||
| + | sortase | ||
| + | reaction took place. <br>Furthermore, we can conclude that the Sortase A7M accepts any primary | ||
| + | amine | ||
| + | as a substrate. However, mass spectrum does not show the ratio of educt and product, which | ||
| + | is | ||
| + | why we cannot estimate whether the turnover is as high as when using a polyG-tag as | ||
| + | substrate. | ||
| + | Additionally this assay confirms our suspicion that the Sortase A7M is stable at 4 °C | ||
| + | and | ||
| + | still functional if stored at said temperature for at least two weeks. <p></p> | ||
| + | <h5>Yield</h5> | ||
| + | <p>For the characterization of Sortase A7M an assay was designed to show the coupling | ||
| + | efficiency | ||
| + | between the TAMRA-LEPTG and the tetrapeptide GGG-Beta-Alanin (GGGβA) catalyzed by the | ||
| + | Sortase. The Sortase reaction was performed for 1h at | ||
| + | 30˚C and was stopped by enzyme separation through centrifugal filtration. For analysis | ||
| + | mass | ||
| + | spectrometry (ESI-MS) was used. The mass spectrometry enables differentiation between | ||
| + | products and educts. It allowed us to make an estimate of the product yield. | ||
| + | The calculated theoretical molecular masses are 1054 g/mol for TAMRA and 1240 g/mol for | ||
| + | TAMRA-LPETGGGβA. Therefore, peaks are expected at mass/n, with n ∈ N. By comparison of | ||
| + | the | ||
| + | number of corresponding peaks, estimation of the product yield is possible as both | ||
| + | molecules | ||
| + | possess the same amount of ionizable groups and thus the difference in the ionizability | ||
| + | of | ||
| + | both molecules is negligible. </p> | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/3/36/T--TU_Darmstadt--results_BC_mass_spectrum.png" | ||
| + | style="max-width:90%" /> | ||
</a> | </a> | ||
<div class="caption"> | <div class="caption"> | ||
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 22 : | |
| − | + | </b> | |
| − | + | Mass spectrum of the sortase-mediated ligation of TAMRA-LPETG and GGGβA | |
| + | showing the difference in height of the educt-peak and the product-peak which can be | ||
| + | used to estimate the yield of our Sortase A7M. | ||
</p> | </p> | ||
</div> | </div> | ||
| + | <p>In <b>Fig. 22</b> the 621.56 peak can be assigned to the TAMRA-LEPTGGGβA and the 528.85 | ||
| + | to | ||
| + | the TAMRA-LPETG. The count ratios of the two molecules mentioned show an excess of the | ||
| + | product. </p> | ||
| − | < | + | <h2>Is Sortase A7M able to attach cargo to P22 coat protein?</h2> |
| − | + | ||
| − | </ | + | |
| − | |||
| − | |||
| − | |||
| − | <img class="img-fluid center" | + | |
| − | + | <p> | |
| + | We performed the linking reaction with CP-LPETGG and GGGG-mCherry as substrates and | ||
| + | applied them to an SDS-PAGE. | ||
| + | We saw products at the expected size (28 kDa + 49 kDa = 77 kDa) thus the | ||
| + | requirement is fulfilled. However, | ||
| + | a lot of additional bands appeared that we did not expect. These bands also appeared | ||
| + | when only Sortase A7M and CP were mixed. | ||
| + | </p> | ||
| + | |||
| + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/1/1d/T--TU_Darmstadt--EnzymeSubstrate1.png" | ||
| + | style="max-width:50%" /> | ||
</a> | </a> | ||
| − | < | + | |
| − | + | ||
| + | <img class="img-fluid center" | ||
| + | src="https://2019.igem.org/wiki/images/7/76/T--TU_Darmstadt--Sortase7Mdiffprot.png" style="max-width:50%" /> | ||
| + | </a> | ||
| + | |||
| + | |||
| + | <p> | ||
<b> | <b> | ||
| − | + | Figure 23: </b> | |
| − | + | <p><b>a)</b> Sortase A7M band is at expected height (17.85 kDa). | |
| − | Sortase | + | The two negative controls containing only GGGG-mCherry (28 kDa) |
| + | and CP-LPETGG (49 kDa) at the expected respective heights. <b>b)</b> Shown are | ||
| + | sfGFP-SP and | ||
| + | CP-LPETGG each incubated with both Sortase A7M and Sortase A5M. | ||
| + | Both gels display multimers when coat and a sortase variant are in a sample | ||
| + | together. | ||
</p> | </p> | ||
| − | |||
| − | <p> | + | <p> |
| − | + | To investigate this issue, we had a look at the | |
| − | </p> | + | literature and found a matching description in the publication of Patterson |
| − | <p> | + | et al.. They performed a similar experiment with P22 capsid proteins and observed |
| − | + | the same multimers in their SDS-PAGEs | |
| − | </p> | + | </sup> |
| − | <p> | + | . Comparing both SDS-PAGEs, we came to the following assumption: |
| − | + | </p> | |
| − | < | + | <p> |
| − | + | Because of the promiscuity of Sortase A7M to accept primary amines as | |
| − | + | substrates, as we discussed previously, the formation of CP multimers occurs, | |
| − | </ | + | unspecifically catalyzed by Sortase A7M. |
| − | + | </p> | |
| − | + | <p> | |
| − | </p> | + | Parallel to these experiments, we successfully modified the exterior of |
| − | + | pre-assembled VLPs <i>in vitro</i> (<a href="http://2019.igem.org/Team:TU_Darmstadt/Project/P22_VLP" | |
| + | target="_blank">VLP assembly</a>). These modified VLPs were homogenous and | ||
| + | overall correctly assembled. <b>Therefore, we conclude that the described multimer | ||
| + | problem only occurs when Sortase A7M encounters free CP.</b> | ||
| + | </p> | ||
| − | |||
| − | < | + | <h2>Does methionine affect Sortase linking?</h2> |
| − | <p> | + | <p> |
| − | + | Sortase A7M preferably attaches N-terminal poly-G to C-terminal LPETGG. However, | |
| − | + | the first amino acid of a protein is methionine (to be specific, formylmethionine in | |
| − | + | bacteria). | |
| − | + | For our constructs that possess N-terminal polyG-tags, we have to ask ourselves the | |
| − | + | question: If the initial | |
| − | + | methionines are not cleaved off after the proteins have been produced, will this | |
| − | + | interfere with the Sortase reaction? | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | < | + | <p> |
| + | To investigate this, we cloned and purified two other proteins: | ||
| + | <b>TVMVsite-GGGG-mCherry</b> and <b>TEVsite-GGGG-sfGFP</b>. | ||
| + | Then we treated these proteins with the respective proteases, resulting in | ||
| + | *GGGG-mCherry and *GGGG-sfGFP. | ||
| + | Following this *GGGG-mCherry was then compared to (M)GGGG-mCherry which we used in | ||
| + | all previous assays. | ||
| + | Assays were also conducted on <b>Fig. 24</b> the processed *GGGG-sfGFP | ||
| + | substrate. | ||
| + | <b>Fig. 24</b> confirmed our assumptions that the unprocessed substrate cannot | ||
| + | be linked to the sorting motif via Sortase A7M. | ||
| + | Subsequently, *GGGG-sfGFP (after protease digest) demonstrate successful linkage via | ||
| + | sortase-mediated ligation. | ||
| + | </p> | ||
| + | <p> | ||
| + | Due to these findings we modified our VLPs with | ||
| + | <a href="https://2019.igem.org/Team:TU_Darmstadt/Project/VLP_Modification" | ||
| + | target="_blank">*GGGG-sfGFP</a>. | ||
| + | </p> | ||
| − | < | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/d/d4/T--TU_Darmstadt--comparison_cleaved_uncleaved_sfGFP.png" | |
| − | + | style="max-width:70%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 24 : | |
| − | + | </b> | |
| − | </p> | + | Sortase-mediated ligation of TAMRA-LPETG and GGGG-sfGFP (with TEV |
| + | cleavage site) one cut with TEV protease and one not. The sample with the unprocessed | ||
| + | substrate shows no increase in RFU. In contrast the processed substrate shows a clear | ||
| + | increase in ΔRFU. After 90 min the reverse reaction begins.The ΔRFU refers to the | ||
| + | respective negative control without each sortase at 514 nm. The mean ΔRFU value of the | ||
| + | duplicates was normalized to zero for better visualization. | ||
| + | </p> | ||
| + | </div> | ||
| − | |||
| + | <p> | ||
| + | We performed FRET-assays with TAMRA-LPETG and either of the following reaction | ||
| + | partners: | ||
| + | </p> | ||
| + | <ul> | ||
| + | <li>(M)GGGG-mCherry, a protein sample that might still carry an N-terminal | ||
| + | methionine</li> | ||
| + | <li>*GGGG-mCherry that does not carry any additional N-terminal residue</li> | ||
| + | </ul> | ||
| + | <p> | ||
| + | Before the FRET-assay was started, we adjusted the mCherry-concentrations of both | ||
| + | fluorescent protein solutions to the same level. To do so, we diluted them until | ||
| + | both showed the same fluorescence at 610 nm. | ||
| + | </p> | ||
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/5/5d/T--TU_Darmstadt--Hannah1.png" |
| + | style="max-width:60%" /> | ||
| + | </a> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/1/17/T--TU_Darmstadt--Hannah2.jpeg" |
| − | + | style="max-width:60%" /> | |
| − | + | </a> | |
| − | + | ||
| − | </ | + | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <p> |
| + | <b> | ||
| + | Figure 25: | ||
| + | </b> | ||
| + | FRET of the sortase reaction connecting TAMRA-LPETG and GGGG-sfGFP mediated by Sortase A7M. The | ||
| + | concentration of the Sortase A7M and TAMRA-LPETG was kept at the same level why the | ||
| + | concentration of sfGFP was either 10,7 µM or 1,4 µM. The graphs show that the reverse | ||
| + | reaction happens earlier if the GGGG-substrate concentration is lower. The ΔRFU refers to the | ||
| + | respective negative control without each sortase at 514 nm. The mean ΔRFU value of the | ||
| + | duplicates was normalized to zero for better visualization. | ||
| + | </p> | ||
| − | <p> | + | <p> |
| − | + | Strikingly, only the (M)GGGG-mCherry construct showed a clear decrease in delta RFU | |
| − | + | after the maximum delta RFU was reached (at about 160 min). | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/f/f4/T--TU_Darmstadt--Hannah3.png" |
| − | + | style="max-width:70%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 26 : | |
| − | </p> | + | </b> |
| + | Sortase-mediated ligation of TAMRA-LPETG and GGGG-mCherry one cut with TVMV protease and one | ||
| + | with a methionine infront of the GGGG-tag. As visible the reverse reaction happens earlier if | ||
| + | the methionine is not cleaved of the GGGG-tag. The ΔRFU is referring to the negative controls | ||
| + | without Sortase A7M at 570 nm. The mean ΔRFU value of the tripicates was normalized to zero | ||
| + | for better visualization. | ||
| + | </p> | ||
| + | </div> | ||
| − | <p> | + | <p> |
| − | + | We assume the following: Although we adjusted the overall mCherry concentration by | |
| − | + | fluorescence, we cannot determine the absolute amount of <b>M</b>GGGG-mCherry in the | |
| − | + | (M)GGGG-mCherry sample. However, if this amount was relatively high, the | |
| − | + | <b>effective substrate concentration</b> that could enter the sortase reaction would | |
| − | + | be low. That is because MGGGG is a worse sortase substrate than GGGG – if any at | |
| − | + | all. If we furthermore consider that a low substrate concentration correlates with a | |
| − | + | faster reverse reaction, we can explain the observed decrease in delta RFU for the | |
| − | + | (M)GGGG-mCherry sample that contrasts the delta RFU trend of the *GGGG-mCherry | |
| − | + | sample. | |
| + | </p> | ||
| + | <p> | ||
| + | On this basis we can assume that a certain, yet unknown portion of the | ||
| + | (M)GGGG-mCherry sample still carries an N-terminal methionine. | ||
| + | </p> | ||
| + | <p> | ||
| + | These FRET-assays let us assume that methionine disturbs or at least interferes with | ||
| + | the sortase reaction mechanism. Indeed, our <b>modeling</b> suggests that | ||
| + | <b>methionine affects the interaction of polyG</b> and the flexible loop near the | ||
| + | active site of Sortase A7M.<a href="http://2019.igem.org/Team:TU_Darmstadt/Model" target="_blank"> | ||
| + | Click here</a> if you want to know more about our modeling | ||
| + | results! | ||
| + | </p> | ||
| − | + | <p> | |
| − | + | This strengthens our hypothesis: If there is any amino acid in front of the | |
| − | </p> | + | poly-glycine sequence, substrate binding to Sortase A7M is negatively influenced. |
| + | </p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <h1>Modeling</h1> |
| − | < | + | <h3>Introduction</h3> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | <p> | + | <p> |
| − | + | In synthetic biology, theoretical models are often used to gain insights, predict | |
| − | + | and | |
| − | + | improve | |
| − | + | experiments. In our project we are modifying Virus-like particles (VLPs) by | |
| − | + | attaching | |
| − | + | proteins to the | |
| − | < | + | surface of the P22 capsid |
| + | <!-- Link zum Background oder Project overview --> through a linker. The linking is | ||
| + | catalyzed using | ||
| + | the enzyme Sortase A7M, which is a calcium-independent mutant of the wild type | ||
| + | Sortase A | ||
| + | <!-- Link zum Sortase Background --> from <i>Staphylococcus aureus</i>. We performed | ||
| + | modeling to predict the unknown structure of the | ||
| + | Sortase A7M, to improve the linker between proteins and therefore optimizing the | ||
| + | modification | ||
| + | efficiency. <br> | ||
| − | < | + | Here <i>Rosetta Comparative Modeling</i> was performed to predict the structure of |
| − | + | Sortase A7M. The generated structure was characterized in terms of the molecular | |
| − | + | mechanisms and binding affinities of ligand peptides using molecular dynamics | |
| − | + | and docking simulations. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| + | <h2>Structure determination</h2> | ||
| − | < | + | <p> |
| − | + | <i>In silico</i> modeling and simulation of proteins requires a 3D structure, | |
| − | + | which can be | |
| − | + | obtained from the <a href="http://www.rcsb.org/" target="_blank">RCSB Protein | |
| − | + | Data | |
| − | + | Bank</a>. However, if no 3D structures are annotated, as it is the case with | |
| − | + | sortase | |
| − | + | A7M, the structure has to be determined by other means. The structure prediction | |
| − | + | of sortase A7M was done using two different approaches. | |
</p> | </p> | ||
| − | |||
| − | + | <h2>RosettaCM</h2> | |
| − | + | ||
| − | + | <h3>Results</h3> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | < | + | <p> |
| − | + | The simulation of our Sortase A7M yielded 15,000 structures which have been compared using the | |
| − | + | Rosetta scoring functions (talaris2013). | |
| − | + | <!-- scoring --> | |
| − | + | From the 15,000 structures generated, we inspected the ten best scoring | |
| − | + | structures. | |
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | < | + | <p> |
| − | + | As can be seen in <b>Fig. 27</b>, the most prominent differences can | |
| − | + | be found in the regions close to the N- and C-terminus. As | |
| − | + | fluctuations in those | |
| − | + | regions are not untypical, we decided to use the best scoring | |
| − | + | structure, candidate S_14771 (<b>Fig.</b> 28), as the input for the | |
| − | + | simulations to follow. | |
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | <img class="img-fluid center" | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/4/40/T--TU_Darmstadt--top10_corporate.png" | |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | </b> | + | Figure 27 : |
| − | + | </b> | |
| + | The structural alignment of the ten best scoring | ||
| + | sortase structures | ||
| + | displaying minor differences with the exception of the C- and | ||
| + | N-terminal | ||
| + | regions. N- and C-terminal regions tend to show strong | ||
| + | fluctuations, thus it is | ||
| + | unsurprising to find the terminal regions to be unaligned | ||
| + | </p> | ||
| + | </div> | ||
| + | |||
| + | |||
| + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/b/b3/T--TU_Darmstadt--s14771.gif" | ||
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 28 : | ||
| + | </b> | ||
| + | Sortase A7M candidate S_14771 created through RosettaCM. | ||
| + | </p> | ||
| + | </div> | ||
| + | |||
| + | <p> In order to evaluate the secondary structure of the Sortase A7M | ||
| + | candidate S_14771 a Ramachandran plot has been created and compared to | ||
| + | the five sortases used as input for the comparitive modeling. | ||
</p> | </p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <img class="img-fluid center" |
| + | src="https://2019.igem.org/wiki/images/2/28/T--TU_Darmstadt--ramachandran_s14711.png" | ||
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <img class="img-fluid center" |
| + | src="https://2019.igem.org/wiki/images/e/ee/T--TU_Darmstadt--ramachandran_five_sortases.png" | ||
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| − | |||
| − | < | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/7/73/T--TU_Darmstadt--Comp_Ramachandran.PNG" | |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | <p> | + | <p> <b>Figure 29</b>: The Ramachandran plot of randomly sampled proteins |
| − | + | and the input structures of the <i>comparative modeling</i> show | |
| − | + | similar secondary structures. Secondary structure analysis of both | |
| − | + | sortase candidates reveals absence | |
| − | + | of secondary structures for the ML candidate. This is not the case | |
| − | + | with candidate S_14771 as the Ramachandran plot shows all relevant | |
| − | + | structures. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | + | <p> | |
| − | + | The Ramachandran plot (<b>Fig. 29</b>) showing α-helices and | |
| − | + | β-sheets is a | |
| − | + | strong indicator of a successful structure determination, as those | |
| − | + | secondary | |
| − | + | structures are crucial for the functionality of sortases. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | < | + | <h3>Conclusion</h3> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| + | <p> | ||
| + | We used monte-carlo simulations | ||
| + | to determine the structure of the mutated transpeptidase Sortase A7M. | ||
| + | As a first investigation of the secondary structure a Ramachandran plot of | ||
| + | candidate <i>S_14771</i> was made. The plot showing secondary structures was a first | ||
| + | indicator | ||
| + | of successful structure prediction. | ||
| + | The approach, using <i>Rosetta Comparative Modeling</i>, yielded | ||
| + | 15,000 | ||
| + | structures scored with the talaris2013 scoring function. The ten | ||
| + | best structures | ||
| + | were aligned and exhibited almost identical secondary structures | ||
| + | (<b>Fig. 27</b>). | ||
| + | The greatest structural differences are present in the N- and | ||
| + | C-terminal regions. Since terminal regions tend to fluctuate more strongly than | ||
| + | non-terminal segments of the protein, we deemed those fluctuations | ||
| + | non-relevant for the proteins functionality. | ||
| + | <br> | ||
| + | Being the best scoring candidate, structure S_14771 was analyzed | ||
| + | structurally using a Ramachandran plot (<b>Fig. 29</b>). The plot shows all the | ||
| + | relevant and typical structures sortases exhibit and serves as an indicator for | ||
| + | a successful structure prediction. | ||
| + | <br> | ||
| + | In the steps to follow, a molecular dynamics (MD) | ||
| + | simulation was performed on both structures. | ||
| + | </p> | ||
| + | <h2>Molecular dynamics</h2> | ||
| − | |||
| − | < | + | <h3>Results</h3> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | <p> | + | <p> |
| − | + | The first possible indicators of a stable protein structure are converging | |
| − | + | root-mean-square deviation (RMSD), | |
| − | + | small root-mean-square | |
| − | + | fluctuation (RMSF) values | |
| − | + | as well as converging radii of gyration. Using the Python software package and | |
| − | + | the module Biotite we calculated | |
| − | + | these quantities and plotted the result for candidate S_14771. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/4/4f/T--TU_Darmstadt--rmsd_s14771.png" |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 30 : | |
| − | + | </b> | |
| − | </ | + | The RMSD is one of three main indicators of a stable |
| + | protein structure of the MD simulation of | ||
| + | S_14771 over the period of 200,000 ps. As time progressed the RMSD | ||
| + | increased with a smaller slope. | ||
| + | The value stabilizes at a time of 110,000 ps and fluctuated around the | ||
| + | value of 6 Å. | ||
| + | </p> | ||
| + | </div> | ||
| − | |||
| − | <p> | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/9/94/T--TU_Darmstadt--gyration_s14771.png" | |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 31 : | |
| − | + | </b> | |
| − | </p> | + | The prominent fluctuations of the residues from ranges |
| + | 105 to 115 might | ||
| + | indicate a binding site or another form of functional structure. The | ||
| + | radius of gyration, just as | ||
| + | the RMSD <b>Fig. 30</b>, stabilizes around a simulation time of of 110,000 ps | ||
| + | and converges towards a value of | ||
| + | 16.7 Å. | ||
| + | </p> | ||
| + | </div> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | <img class="img-fluid center" | + | |
| − | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/f/f4/T--TU_Darmstadt--rmsf_s14771.png" | |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 32 : | |
| − | + | </b> | |
| − | + | The fluctuations | |
| − | + | (RMSF) of most residues appear insignificant compared to the first, the | |
| − | + | last residues and | |
| − | + | the residues close to residue 110 . Typically the N- and C-terminus tend | |
| − | + | to fluctuate more intensively due to the lack of | |
| − | + | stabilizing structures. The prominent fluctuations in the range of | |
| − | + | residue 105 to 115 | |
| − | + | can indicate a binding site or another form of functional structure. | |
| + | </p> | ||
| + | </div> | ||
| + | |||
| − | < | + | <p> |
| − | + | Typical RMSDs and radii of gyration converge towards a value dependent on the | |
| − | + | size of the | |
| − | + | protein. Convergence of those quantities can be interpreted as a stable state of | |
| − | + | the protein | |
| − | + | structure. As it can be seen in <b>Fig. 30</b> and <b>Fig. 31</b> both the RMSD and | |
| − | + | the radius of | |
| − | + | gyration | |
| − | + | stabilize at the same time as the simulation reaches 110,000 ps (110 ns), | |
| − | + | suggesting a now | |
| − | + | stabilized structure of candidate S_14771 solvated in water. Another indicator | |
| − | + | of a | |
| + | functional protein is the RMSF. Instead of being averaged over all atoms, the | ||
| + | RMSF is | ||
| + | averaged over time with respect to each amino acid. It provides insights in both | ||
| + | protein | ||
| + | stability and functionality. Fig <b>Fig. 32</b> reveals the RMSF of residues 105 to | ||
| + | 115 to | ||
| + | be | ||
| + | significantly higher than that of other residues. This hints at the presence of | ||
| + | a | ||
| + | functional unit along these residues. As commented on in the section | ||
| + | describing our structure prediction approaches, the N- | ||
| + | and C-terminal regions tend to fluctuate more strongly as a result of the | ||
| + | absence of | ||
| + | stabilizing structures. | ||
</p> | </p> | ||
| − | |||
| − | < | + | <p> |
| − | + | To further analyse the potential binding site, we performed a | |
| − | + | <i>Principle Component Analysis</i>. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| + | <h3>Principle component analysis</h3> | ||
| − | + | <p> | |
| − | + | To analyze our system further Principle Component Analysis (PCA) was performed | |
| − | + | using GROMACS. By applying PCA to a protein it is possible to gain insights into the | |
| − | + | relevant | |
| − | + | vibrational motions and thereby the physical mechanism of the protein. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/d/db/T--TU_Darmstadt--modes_s14771.gif" |
| − | + | style="width:40%"> | |
| − | + | <p><b>Animation 33: </b> A Principle Component Analysis of a fast (blue) and a | |
| − | + | slow (red) mode showing the most prominent movements of the Cα-chain | |
| − | + | of candidate S_14771. Both modes show movement of the β6/β7 | |
| − | + | loop consisting of residues 105 to 115 towards the active site . Thus we can | |
| − | + | assume that the closing β6/β7 loop is involved in the reaction | |
| − | + | mechanism. | |
| − | + | </p> | |
| − | + | ||
| − | </p> | + | |
| − | <p> | + | <p> |
| − | + | The results from the Principle Component Analysis of candidate S_14771 | |
| − | + | (<b>Amination 33</b>) show a movement of the residues 105 to 115 towards the active | |
| − | + | site, supporting our theory that residues 105 to 115 are important for the | |
| − | + | reaction mechanism. Since the most relevant eigenvector (red), which shows the most | |
| − | + | relevant | |
| − | </p> | + | movement of the sortase, moves further towards the active site, it is possible |
| + | that the β6/β7 loop either closes the binding site of the ligand | ||
| + | peptides or even transports one peptide towards the other. | ||
| + | </p> | ||
| − | <h3> | + | <h3>Conclusion</h3> |
| − | <p> | + | <p> |
| − | + | Candidate S_14771 that was generated using <i>RosettaCM</i> appears to be a fitting | |
| − | + | candidate not | |
| − | </p> | + | only due to successful analyses, but also due to the residues of the active site |
| + | being in close proximity. | ||
| + | Not the RMSF but also Principle Component Analysis | ||
| + | indicate that residues 105 to 115 forming the β6/β7 loop | ||
| + | are crucial for the molecular mechanism of our model. | ||
| + | </p> | ||
| + | <h2>Docking</h2> | ||
| − | + | <p> | |
| − | + | Now that the binding site of the Sortase had been found, the peptide ligand | |
| − | + | needed to be inserted into the binding site to create a peptide-protein complex. | |
| − | + | The procedure of choice | |
| − | + | for the introduction of a ligand into the binding site of a protein is called | |
| − | + | <i>docking</i>. In the | |
| − | + | following sections, we will present the protocol and methods we used as well as | |
| − | + | the results they yielded. | |
| − | + | </p> | |
| − | + | ||
| − | < | + | <h3>Results</h3> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | <p> | + | <p> |
| − | + | For sequences MGGGGPPPPPP(M-polyG), GGGGPPPPPP(polyG) and PPPPPPLPETGG(LPETGG) | |
| − | + | 50,000 structures have been created and clustered. | |
| − | + | After the clustering the sample consisted of 100 structures of docked complexes. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/7/78/T--TU_Darmstadt--dock_lpetgg.png" |
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 34 : | ||
| + | </b> | ||
| + | The three best scoring structures (total score, interface | ||
| + | score, reweighted score) of the LPETGG-tag are shown. Only two results are | ||
| + | visible as the best reweighted score candidate is identical to the best | ||
| + | interface score candidate. The reacting section of the LPETGG-tag namely | ||
| + | glycine is colored yellow as is the active site. The glycin of both ligand | ||
| + | peptides is facing the active site. | ||
| + | </p> | ||
| + | </div> | ||
| − | <p> | + | <p> |
| − | + | Analysis of the scores has shown a similar score for all the three dockings. The | |
| − | + | best scoring results of the LPETGG docking show a tendency of the glycines to | |
| − | + | face the active site while also being in close proximity to the active site. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | < | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/8/8d/T--TU_Darmstadt--dock_polyg.png" |
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 35: | ||
| + | </b> | ||
| + | The three best scoring structures (total score, | ||
| + | interface score, reweighted score) of the polyG peptide are shown. Only | ||
| + | two results are visible as the best reweighted score candidate is | ||
| + | identical to the best interface score candidate. Instead of facing the | ||
| + | active site (yellow) the reacting glycines (yellow) appear to interact | ||
| + | with the β6/β7 loop of the sortase. | ||
| + | </p> | ||
| + | </div> | ||
| − | <p> | + | <img class="img-fluid center" src="https://2019.igem.org/wiki/images/9/92/T--TU_Darmstadt--dock_mpolyg.png" |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 36: | |
| − | + | </b> | |
| − | </p> | + | The three best scoring structures (total score, |
| + | interface score, reweighted score) of the poly-g peptide are shown. Only | ||
| + | two results are visible as the best reweighted score candidate is | ||
| + | identical to the best interface score candidate. | ||
| + | Concerning the M-poly-G peptide no uniform directional orientation can | ||
| + | be observed. | ||
| + | The structure with the best interface score (light blue) is oriendted | ||
| + | towards the loop while the structure with the best total/reweighted | ||
| + | (dark blue) is oriented towards the β-sheets. | ||
| + | </p> | ||
| + | </div> | ||
| − | < | + | <p> |
| + | <b>Fig. 34</b> shows the docking result of the LPETGG peptide to | ||
| + | the sortase. The results shown are the best scoring structures of the clustering | ||
| + | with respect to the total score, interface score and reweighted score. As the | ||
| + | best scoring structure is the same for the total score and the reweighted score | ||
| + | only two peptides are shown. This also applies to <b>Fig. 35</b> and <b>Fig. 36</b>. | ||
| + | For both | ||
| + | results the reacting glycin residues (yellow) are facing the active site. | ||
| + | Additionally, the same residues are in close proximity to the active site. | ||
| + | </p> | ||
| − | <p> | + | <p> |
| − | + | The figures <b>Fig. 35</b> and <b>Fig. 36</b> show the docking of the both polyG and | |
| − | + | M-polyG. While polyG | |
| − | + | results align well and seem to be interacting with the β6/β7 loop | |
| − | + | rather than with the active site, this does not seem to be the case for M-polyG. | |
| − | + | Instead of both structures interacting with the β6/β7 loop or | |
| − | + | active site one (best interaction score; dark blue) interacts with the | |
| − | + | β6/β7 loop and the other (best reweighted/total score; light | |
| − | + | blue-gray) appears to interact with the active site. | |
| − | </p> | + | </p> |
| − | < | + | <img class="img-fluid center" |
| + | src="https://2019.igem.org/wiki/images/7/76/T--TU_Darmstadt--dock_zoom_active.png" | ||
| + | style="max-width:40%" /> | ||
| + | </a> | ||
| + | <div class="caption"> | ||
| + | <p> | ||
| + | <b> | ||
| + | Figure 37: | ||
| + | </b> | ||
| + | The close up of the M-polyG peptide (best | ||
| + | total/reweighted score) indicates an interaction of methionine with | ||
| + | arginine<sub>139</sub> and cysteine<sub>126</sub>. | ||
| + | </p> | ||
| + | </div> | ||
| − | < | + | <img class="img-fluid center" |
| − | + | src="https://2019.igem.org/wiki/images/4/48/T--TU_Darmstadt--dock_zoom_loop.png" | |
| − | + | style="max-width:40%" /> | |
| − | + | </a> | |
| − | + | <div class="caption"> | |
| − | + | <p> | |
| − | + | <b> | |
| − | + | Figure 38: | |
| − | + | </b> | |
| − | + | Methionine of the result with the best interface score | |
| − | + | interacted with the β6/β7 loop rather than the active | |
| − | + | site. Still the reactive glycine residues appear to be bound to the | |
| − | + | β6/β7 loop. | |
| − | + | </p> | |
| − | + | </div> | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | < | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | <p> | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| + | <p> | ||
| + | As can be seen in <b>Fig. 37</b> visualizing the result of the the docking | ||
| + | simulation | ||
| + | (total/reweighted score) suggests an interaction of methionine and two of the | ||
| + | active sites namely arginine<sub>139</sub> and cysteine<sub>126</sub>. | ||
| + | <b>Fig. 38</b> shows the interaction of M-polyG with the β6/β7 loop. | ||
| + | The glycines still interact with the β6/β7 loop. | ||
| + | Instead of binding above the β6/β7 loop, which is the case for | ||
| + | polyG as illustrated in <b>Fig. 36</b>, | ||
| + | the interaction seems to be influenced by methionine. By interacting with the | ||
| + | residues in the β-helix | ||
| + | methionine could potentially hinder binding of glycine to the | ||
| + | β6/β7 loop by partial | ||
| + | immobilization of the peptide. Overall peptide binding and orientation is less | ||
| + | uniform compared | ||
| + | polyG without the leading methionine, which could be an indicator of lesser | ||
| + | binding affinity of M-PolyG towards | ||
| + | the β6/β7 loop. | ||
| + | </p> | ||
| − | <h3> | + | <h3>Conclusion</h3> |
| − | <p> | + | <p> |
| − | + | To computationally investigate binding affinities of the polyG and M-polyG as | |
| − | + | well as the LPETGG tags we performed | |
| − | + | docking simulations using the <i>Rosetta FlexPepDock</i> application. We used a | |
| − | </p> | + | modified version of the recommended |
| + | protocol as the modified version was easier to automate and served our purpose | ||
| + | better than the standard protocol. | ||
| + | From the calculated scores only, we could not see a difference in binding | ||
| + | affinities. | ||
| + | Thus, we inspected the best scoring | ||
| + | structures regarding the total score, the interface score and the reweighted | ||
| + | score using PyMOL. | ||
| + | Since the best structures with respect to total score and reweighted score were | ||
| + | the same for all simulations, | ||
| + | only two structures have been inspected per run. A polyproline tag was appended | ||
| + | to all the peptides to simulate | ||
| + | the modification of the VLPs with a small peptide. | ||
| + | <!-- GRoß helices etc erwähnen als begründung --> | ||
| + | </p> | ||
| − | < | + | <p> |
| − | + | As expected, the results showed that for LPETGG, the glycines of both peptides | |
| − | + | oriented towards the active site. | |
| − | + | This is unsurprising as peptides with the sequence LPXTGG are known to be | |
| − | + | substrate of the Sortase. It was more surprising to | |
| − | + | see the polyG tag oriented away from the active site since polyG also is a known | |
| − | + | substrate of the sortase. Both polyG peptides | |
| − | + | were facing the β6/β7 loop (residues 105 to 115) uniformly and | |
| − | + | appeared to be interacting with it. The M-polyG peptides did not | |
| − | + | show a uniform orientation or interaction scheme. On one hand the visualization | |
| − | + | of the best result concerning the total and reweighted | |
| − | + | score has shown interaction of methionine with the cysteine<sub>126</sub> and | |
| − | + | arginine<sub>139</sub>, two residues of the active | |
| − | + | site. On the other hand, the visualization of the best result with respect to | |
| + | the interface score shows the M-polyG facing the mobile β6/β7 | ||
| + | loop. | ||
| + | In contrast to the polyG peptide the lacking the methionine, the M-polyG peptide | ||
| + | is pulled down below the β6/β7 loop by the methionine interacting | ||
| + | with one of the β-sheets leading to the active site. This is not the case | ||
| + | with the polgG results, which lie aligned in one plane | ||
| + | with the β6/β7 loop. | ||
</p> | </p> | ||
| − | |||
| − | < | + | <h2>Modeling Conclusion</h2> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | </ | + | |
| − | < | + | <p> |
| − | + | For our project it was key to understand and characterize Sortase A7M. | |
| − | + | As there is no annotated 3D structure for this specific Sortase, an <i>in | |
| − | + | silico</i> structure determination | |
| − | + | was performed. Comparative modeling with Rosetta produced valid structures. We used | |
| − | + | the best structure, candidate S_14771, for extensive characterization. | |
| − | + | We evaluated the model with regard to its secondary structure using Ramachandran | |
| − | + | plots. The Ramachandran plot suggested plausible secondary structures. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | + | <p> | |
| − | + | Molecular Dynamics simulations were used to investigate stability and dynamic | |
| − | + | properties of the candidate. | |
| − | + | The RMSD and radius of gyration stabilized over the course of the simulation, a | |
| − | + | first indicator of an equilibrated structure. | |
| − | + | Interestingly, RMSF analysis showed strond fluctuations of residues 105 to 115. | |
| − | + | We further investigated this by performing | |
| − | + | Principle Component Analysis. Doing so, we extracted the principle movements of | |
| − | + | the model. We could observe movement | |
| − | + | of the β6/β7 loop towards the active site, suggesing the presence | |
| − | + | of a binding site. | |
| − | + | Consequently, we performed docking simulations. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | <p> | + | <p> |
| − | + | FlexPepDock was used to conduct the docking simulations with target peptides. | |
| − | + | Each run yielded 50,000 structures. | |
| − | + | In multiple steps we reduced the amount of complexes to 100 clusters with | |
| − | + | respect to total, reweighted and interface score. | |
| − | + | We extracted the best scoring complexes and investigated interactions. | |
| − | + | </p> | |
| − | + | ||
| − | + | ||
| − | </p> | + | |
| − | <p> | + | <p> |
| − | + | For LPETGG we observed a uniform binding to the active site, fullfilling our | |
| − | + | expectation. | |
| − | + | Strikingly, polyG appeared to bind to the β6/β7 loop in a uniform | |
| − | + | manner. | |
| − | + | As it is know from literature polyG is a functioning ligand of sortase. | |
| − | + | Supported by literature and our data, <b>we postulate the following | |
| − | + | mechanism</b>: | |
| − | + | the β6/β7 loop transports bound polyG towards the active site of | |
| − | + | Sortase A7M, thereby lowering the activation energy of the linking reaction. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | |||
| − | + | <p> | |
| − | + | As the theory is neither backed up by nor contradicts experimental data, further | |
| − | + | research is required. | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
</p> | </p> | ||
| − | + | <h2>References</h2> | |
| + | <p>For our references, please have a look at our <a href="http://2019.igem.org/Team:TU_Darmstadt">wiki</a>.</p> | ||
| − | < | + | </html> |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ==Contribution: XHD-Wuhan-Pro-China 2021== | |
| − | + | ===literature 1=== | |
| − | + | Bacteria display a variety of proteins on their surface that to enable them to effectively interact with their environment. Gram-positive bacteria use sortase cysteine transpeptidase enzymes to covalently attach proteins to their cell wall, and to assemble pili. Sortases in pathogenic bacteria are frequently important virulence factors, as many of the proteins that they display have key roles in the infection process, such as promoting bacterial adhesion, nutrient acquisition, and the evasion and suppression of the immune response (Cascioferro, Totsika, & Schillaci, 2014; Schneewind & Missiakas, 2012, 2014; Siegel, Liu, & Ton-That, 2016; Spirig, Weiner, & Clubb, 2011). As a result, a significant amount of effort has been put forth to elucidate the mechanism of sortase-mediated catalysis and to discover small-molecule sortase inhibitors that could function as potent antiinfective agents (Bradshaw et al., 2015; Cascioferro et al., 2014; Clancy, Melvin, & McCafferty, 2010; Maresso & Schneewind, 2008; Suree, Jung, & Clubb, 2007). Moreover, sortases have been developed into valuable biochemical reagents to ligate distinct biomolecules together via a covalent peptide bond. This in vitro transpeptidation activity has been harnessed for a variety of useful applications, including among others, covalently attaching proteins to cells, attaching fluorophores or drugs to antibodies, cyclizing proteins, and immobilizing peptides on solid surfaces (Antos, Truttmann, & Ploegh, 2016; Popp & Ploegh, 2011; Ritzefeld, 2014; Schmidt, Toplak, Quaedflieg, & Nuijens, 2017; Schmohl & Schwarzer, 2014; Voloshchuk, Liang, & Liang, 2016). | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | Sortases perform two distinct functions in bacteria: (1) attach proteins directly to the cell wall or (2) assemble pili, long proteinaceous fibers that project from the microbial surface (Fig. 1). Both reactions are mechanistically related and operate on secreted proteins that contain a C-terminal cell wall sorting signal (CWSS). The CWSS contains a five-residue sortase recognition motif, frequently LPXTG, that is followed by a hydrophobic domain and a positively charged cytoplasmic anchor that retains the protein substrate in the membrane (Schneewind, Model, & Fischetti, 1992). The Sortase A enzyme from Staphylococcus aureus (SaSrtA) has been studied in detail and is archetypal (Mazmanian, Liu, Ton-That, & Schneewind, 1999; Ton-That, Liu, Mazmanian, Faull, & Schneewind, 1999). SaSrtA attaches surface proteins to the cell wall by recognizing an LPXTG motif within the CWSS of its protein substrate (Fig. 2A). Catalysis begins when SaSrtA’s active site cysteine residue nucleophilically attacks the carbonyl carbon in the peptide bond between the Thr and Gly residues in the sorting signal (Fig. 2A, step 1) (Clancy et al., 2010; Connolly & Clubb, 2005). This generates a tetrahedral intermediate that quickly collapses to form a semi-stable thioacyl intermediate in which sortase is covalently linked via its cysteine residue to its protein substrate (Fig. 2A, step 2). SaSrtA then recognizes a second substrate, the cell wall precursor, lipid II (Fig. 2A, step 3) (Perry, Ton-That, Mazmanian, & Schneewind, 2002; Ruzin et al., 2002) and catalyzes a reaction in which the N-terminal primary amine group within the cross-bridge peptide nucleophilically attacks the carbonyl carbon atom within the thioacyl bond. This second transient tetrahedral intermediate resolves into the protein–lipid II product in which the components are joined via a peptide bond (Fig. 2A, step 4) (Perry et al., 2002; Schneewind, Fowler, & Faull, 1995; Ton-That, Faull, & Schneewind, 1997). The lipid II-linked protein is then incorporated into the peptidoglycan via the transpeptidation and glycosylation reactions that synthesize the cell wall. In contrast to attaching proteins to the cell wall, a second type of sortase, frequently called “pilin polymerases,” construct bacterial pili by polymerizing pilin protein subunits (Fig. 2B) (Hendrickx, Budzik, Oh, & Schneewind, 2011; Kline, Dodson, Caparon, & Hultgren, 2010; Mandlik, Swierczynski, Das, & Ton-That, 2008; Siegel et al., 2016; Spirig et al., 2011; Ton-That & Schneewind, 2003). These pilin-assembling enzymes employ a similar transpeptidation reaction as SaSrtA, but instead of using lipid II as a nucleophile to attach proteins to the cell wall, a lysine amino group located within a protein pilin subunit is used as a secondary substrate to attack the sortase–protein thioacyl intermediate (Fig. 2B, steps 3 and 4). This reaction constructs pili by covalently linking protein subunits together via lysine–isopeptide bonds. Both types of sortase-catalyzed processes occur on the extracellular membrane, where the enzyme and its substrate are membrane associated (Cozzi et al., 2011; Spirig et al., 2011; Wu et al., 2012). | |
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | ||
| − | + | [[File:T-- XHD-Wuhan-Pro-China--2.jpg|750px|]] | |
| − | + | Sortase enzymes attach proteins to the cell wall and assemble pili. (A) Overview of anchoring and pilus assembly reactions. A protein that is to be displayed (blue) contains an N-terminal secretion signal and a C-terminal cell wall sorting signal (CWSS). The CWSS contains an LPXTG-like sorting signal sequence that is processed by the sortase, a nonpolar polypeptide segment (black), and a C-terminal segment of positively charged residues (+). After secretion through the Sec translocon, the protein remains embedded in the lipid bilayer via the nonpolar segment within the CWSS. The sortase enzyme then cleaves between the threonine and glycine residues to form a sortase–protein thioacyl intermediate in which the active site cysteine is covalently linked to the carbonyl carbon atom of the threonine. There are two basic types of sortases: (1) cell wall anchoring sortases that attach protein to the crossbridge peptide of the cell wall and (2) pilin polymerase sortases that covalently link pilin subunits together via lysine–isopeptide bonds. In both cases, the enzymes function as transpeptidases. Some sortases are capable of performing both functions, attaching proteins to the cell wall and polymerizing pili. | |
| − | + | ||
| − | + | ||
| + | [[File:T-- XHD-Wuhan-Pro-China--3.jpg|750px|]] | ||
| − | + | Mechanism of cell wall protein anchoring and pilus assembly. Sortases perform two basic functions in bacteria: (1) attach proteins to the cell wall and (2) join proteins together to construct pili. (A) In the cell wall anchoring reaction, the sortase and substrate are both membrane bound. The reaction occurs via four distinct steps. Sortase first recognizes a sorting signal motif within the CWSS and nucleophilically attacks the threonine’s carbonyl carbon atom via its active site cysteine residue (for demonstration purposes the LPXTG sorting signal recognized by class-A type enzymes is shown, step 1). The LPXTG sorting signal is then cleaved to produce a sortase–substrate thioacyl intermediate (step 2). Next, the crossbridge peptide from a lipid II molecule nucleophilically attacks the thioacyl intermediate (step 3). Lastly, a new peptide bond is formed between the lipid II molecule and surface protein to produce a protein–lipid II intermediate that is incorporated into cell wall by the transglycosylation and transpeptidation reactions that synthesize the peptidoglycan (step 4). (B) In the pilus assembly reaction, steps 1–2 produce a sortase–substrate thioacyl intermediate, similar to the cell wall anchoring reaction. In this reaction, the sortase recognizes a pilin protein that contains a CWSS. However, a lysine residue within the pilin motif from an adjacent pilin protein performs the nucleophilic attack on the thioacyl intermediate (step 3). A new protein–protein isopeptide bond is formed that covalently links the pilin proteins (step 4). This assembly process is repeated to build an isopeptide-linked pilus shaft that contains multiple pilin proteins. Depending on the type of pilus, distinct tip and base pilin proteins can be located at the ends of the pilus shaft, which are incorporated through a similar mechanism and involve covalent linkages via lysine-derived isopeptide bonds. Finally, the intact pilus is attached to the cell wall via sortase-catalyzed attachment of the pilus to lipid II, similar to cell wall protein display. Some sortases are capable of performing both functions, attaching proteins to the cell wall and functioning as pilin polymerases. | |
| + | |||
| + | At present, 3330 gene sequences encoding sortase enzymes have been identified within 1098 species of bacteria (Finn et al., 2016). Sortases are primarily found in Gram-positive bacteria, but are also present to a lesser extent in some species of Gram-negative and archaebacteria. Based on their primary sequences, most sortase homologs can be classified into six distinct subfamilies, called class A–F enzymes (Dramsi, Trieu-Cuot, & Bierne, 2005; Spirig et al., 2011). Class A, B, and D enzymes are prevalent in bacteria within the Firmicutes phylum, while class E and F enzymes predominate in Actinobacteria. Class C enzymes are found in both Firmicutes and Actinobacteria. Similar to SaSrtA (a class A enzyme), all sortases contain a His–Cys–Arg catalytic triad (Ilangovan, Ton-That, Iwahara, Schneewind,& Clubb, 2001; Marraffini, Ton-That, Zong, Narayana, & Schneewind, 2004; Ton-That, Mazmanian, Alksne, & Schneewind, 2002), and their primary sequences harbor a highly conserved TLXTC motif that contains the catalytically essential cysteine residue (Clancy et al., 2010). All sortases characterized to date catalyze a transpeptidation reaction that joins an LPXTG-like sorting signal within the CWSS of their protein substrate to an amino nucleophile. However, their sorting signal and nucleophile substrate specificities can vary substantially. These distinct specificities enable microbes to utilize more than one type of sortase to elaborate their surface, with each sortase operating nonredundantly to display or assemble distinct proteins on the cell surface (Comfort & Clubb, 2004; Pallen, Lam, Antonio, & Dunbar, 2001). | ||
| + | |||
| + | A number of excellent reviews have been written that describe the overall function of sortases in bacteria, their development as biochemical reagents, and efforts to discover therapeutically useful sortase inhibitors (Antos et al., 2016; Bradshaw et al., 2015; Cascioferro et al., 2014; Clancy et al., 2010; Maresso & Schneewind, 2008; Popp & Ploegh, 2011; Ritzefeld, 2014; Schmidt et al., 2017; Schmohl & Schwarzer, 2014; Schneewind & Missiakas, 2012, 2014; Siegel et al., 2016; Spirig et al., 2011; Suree et al., 2007; Voloshchuk et al., 2016). In this chapter, we review what is known about their atomic structures and the molecular basis of substrate recognition and catalysis. | ||
| + | |||
| + | ===Reference=== | ||
| + | Ge Y, Chen L, Liu S, Zhao J, Zhang H, Chen PR. Enzyme-Mediated Intercellular Proximity Labeling for Detecting Cell-Cell Interactions. J Am Chem Soc. 2019 Feb 6;141(5):1833-1837. doi: 10.1021/jacs.8b10286. Epub 2019 Jan 29. PMID: 30676735. | ||
| + | |||
| + | ===Literature 2=== | ||
| + | Strategies for site-specific protein modification are highly desirable for the construction of conjugates containing non-genetically-encoded functional groups. Ideally, these strategies should proceed under mild conditions, and be compatible with a wide range of protein targets and non-natural moieties. The transpeptidation reaction catalyzed by bacterial sortases is a prominent strategy for protein derivatization that possesses these features. Naturally occurring or engineered variants of sortase A from Staphylococcus aureus catalyze a ligation reaction between a five-amino-acid substrate motif (LPXTG) and oligoglycine nucleophiles. By pairing proteins and synthetic peptides that possess these ligation handles, it is possible to install modifications onto the protein N- or C-terminus in site-specific fashion. As described in this unit, the successful implementation of sortase-mediated labeling involves straightforward solid-phase synthesis and molecular biology techniques, and this method is compatible with proteins in solution or on the surface of live cells. | ||
| + | |||
| + | ===Reference=== | ||
| + | Antos JM, Ingram J, Fang T, Pishesha N, Truttmann MC, Ploegh HL. Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation. Curr Protoc Protein Sci. 2017 Aug 1;89:15.3.1-15.3.19. doi: 10.1002/cpps.38. PMID: 28762490; PMCID: PMC5810355. | ||
| + | |||
| + | |||
| + | |||
| + | |||
| + | <!-- Add more about the biology of this part here | ||
| + | ===Usage and Biology=== | ||
Latest revision as of 14:02, 19 October 2021
Sortase A7M (Ca2+-independent variant)
Profile
| Name | Sortase A7M |
| Base pairs | 450 |
| Molecular weight | 17.85 kDa |
| Origin | Staphylococcus aureus, synthetic |
| Properties | Ca2+-independent, transpeptidase, linking sorting motif LPXTG to poly-glycine Tag |
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21INCOMPATIBLE WITH RFC[21]Illegal XhoI site found at 445
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000COMPATIBLE WITH RFC[1000]
Structure

Figure 1 : Modeled 3D-Structure of Sortase A7M. To find out more, visit our modeling page. For the PDB file of our model click here.
Usage and Biology
Transpeptidase: Sortase
Sortases belong to the class of transpeptidases and are mostly found in gram-positive bacteria.
The high rate of resistance to several antibiotics targeting gram-positive bacteria is also based on the
property of this enzyme class. Sortases can non-specifically attach virulence and
adhesion‐associated proteins to the peptidoglycans of the cell-surface.
In general, sortases are divided into six groups (A-F) that have slightly different properties and
perform three tasks in cells. Group A and B attach proteins to the cell-surface while Group C and D help
building pilin-like structures. Group E and F are not properly investigated yet which is why their exact
function is not known.
For our project we are especially interested in the sortases of the group A since they
covalently attach various proteins or peptides on the cell membrane as long as their targeting
motif is at the C-terminus of the corresponding protein. In comparison to other transpeptidases
Sortase A has the advantage that it is rather stable regarding variations in pH
Sortase A catalyzes the formation and cleavage of a peptide bond between the C-terminal
LPXTG amino acid motif and an N-terminal poly-glycine motif. The enzyme originates from
Staphylococcus aureus and is able to connect any two proteins as long as they possess those
matching target sequences. In the pentapeptide motif LPXTG, X can be any amino acid except cysteine.
Sortase A is rather promiscuous with regard to the amino acid sequence directly upstream of
this motif, a fact that makes it optimal for labeling applications. Even better, amino acids C-terminal
of the poly-glycine motif are not constrained to a certain sequence.
Reaction
To better understand how the enzymatic reaction works it is necessary to look at the crystal structure of Sortase A. The enyzme consists of an eight-stranded β‐barrel fold structure. The active site is hydrophobic and contains the catalytic cysteine residue Cys184 as well as a key histidine residue H120 that can form a thiolate-imidazolium with the neighboring cysteine. An additional structural property that also other sortases show is the calcium binding site formed by the β3/β4 loop. The binding of a calcium ion slows the motion of the active site by coordinating to a residue in the β6/β7 loop. This helps binding the substrate and increasing the enzymatic activity nearly eightfold. When a substrate gets into the active site, the cysteine attacks the amide bond between the threonine and the glycine in the LPXTG motif. After this the protonated imidazolium serves as an acid for the departing glycine with unbound NH2 of the former amide bond while the rest of the motif is bound to the cysteine residue. Another glycine nucleophile is then necessary in its deprotonated form to attack the thioester and re-establish an amide bond at the LPET-motif. This reaction is dead-ended if the used nucleophile is water. Due to the fact that the mechanism is based on protonated forms of the catalytic residues the reaction is quite pH-dependent. Although the Sortase A in general is relatively stable between pH 3 and 11 the reaction works best around pH 8.
Sortase variants
Due to the fact that the wildtype Sortase A shows rather slow kinetics, a pentamutant has been developed (Sortase A5M). This version of the enzyme carries mutations in P94R/D160N/D165A/K190E/K196T which lead to a 140- fold increase in activity. Thereby, reaction rates are improved even at low temperature, however, Sortase A5M is still Ca2+-dependent. This dependence interferes with potential in vivo usage, as the concentrations of calcium in living cells can vary considerably. Hence a sortase mutant that acts across high differences in calcium concentrations or even works completely Ca2+-independently would be required for in vivo applications of sortase. To attain a high yield enzyme which is also calcium-independent Ca2+-independent mutations were combined with the Sortase A5M resulting in Sortase A7 variants such as the Sortase A7M. The newly achieved calcium-independence of these variants enable sortase applications not only in vitro but in vivo as well.
Sortase A7M
For our project we chose to work with this optimized Sortase A7M. Its size is about 17.85 kDa and it has been shown to be stable for several weeks in the fridge at 4 °C. It also possesses the same properties of pH stability like other sortases but comes with the advantage of being calcium independent. "Sortagging" applications have included the cyclization of proteins and peptides , modification and labeling of antibodies and the synthesis of protein conjugates with drugs, peptides, peptide nucleic acids and sugars.Moreover it poses a lot of advantages for the binding of two proteins in vivo since it has relatively small tags which avoids putting too much metabolic burden on the cells when expressing the proteins of interest. This also avoids disturbing the folding of the proteins of interest and the later biological functions since the Sortase A7M is able to work under physiological conditions. Other methods like the intein- based labeling of surfaces require large fusion-proteins with the intein domain which puts stress on the living cells and might cause folding and solubility issues. Another application for sortase-mediated systems is the anchoring of proteins on the cell wall of gram-positive bacteria which can be used for display of heterologous proteins. It is also possible to attach non-biological molecules to the respective tag. The accessibility and flexibility determine the ability of a sortase enzyme to recognize the sorting motif and catalyzing the transacylation.
Methods
Cloning
The methods used for cloning of the different mutants of the sortase were restriction and ligation via NdeI and SalI and Gibson assembly. The vector posesses a kanamycin resistance and the srta7m is controlled through a T7 promoter, which can be induced with IPTG. Sortase A7M is controlled by the same T7 promoter. The product was checked via sequencing.
Expression and purification
After successfully transforming our sortase genes in BL21 cells, we inoculated 100 mL overnight cultures, with the respective antibiotic. The next day 1 L cultures were inoculated with the overnight culture to reach OD600 = 0.1. Subsequently the cultures were incubated under constant shaking at 37 °C until they reached OD600 = 0.6. At OD600 = 0.6 the cultures were induced with 0.5 mL of 1 M Isopropyl-β-D-thiogalactopyranosid (IPTG). The gene expression was performed at 30 °C under constant shaking overnight. After expression of Sortase A7M in BL21 cultures the cells were crushed via EmulsiFlex (Avestin) and proteins were purified through affinity chromatography via Fast Protein Liquid Chromatography (FPLC) with the ÄKTA pure (GE Healthcare, Illinois, USA). His-Tag was used for purification of Sortase A7M and Sortase A (Stockholm) and Strep-Tag II was used for purification of Sortase A5M.
SDS-Page
To verify the successful production of of Sortase A7M and others SDS-PAGEs were performed. The resulting bands were compared to the molecular weight of the different sortase variants. Also, SDS-PAGEs were completed to verify enzymatic activity in assays prior to measuring sortase properties via Fluorescence Resonance Energy Transfer (FRET).
Flourescence Resonance Energy Transfer (FRET)
To determine the kinetics of our transpeptidase variants, FRET assays were performed in 384 well-plates (dark) using a Tecan plate reader. A FRET relies on the phenomenon that an excited fluorophore (donor) transfers energy to another fluorophore (acceptor), thereby exciting it. This process only works if both fluorescent molecules are in close proximity and depends on the FRET-Pair. By transferring the energy from donor to acceptor, the donor's emission is reduced and the intensity of the acceptors emission is increased . The efficiency depends on the distance between the fluorophore, the orientation and the spectral characteristics . You can see the principle of FRET in Fig. 2.
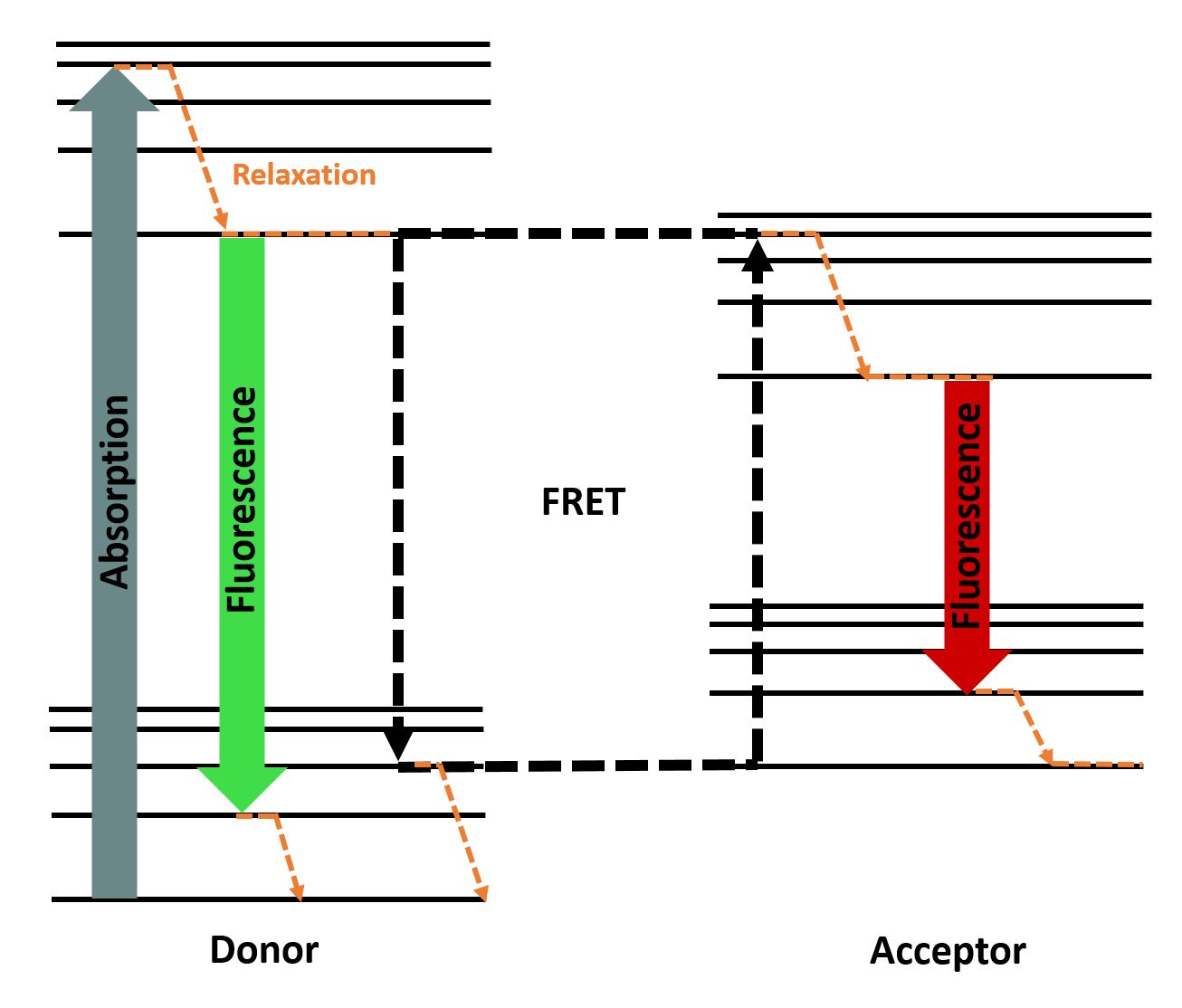
Figure 2 : Jablonski diagram showing the energy transfer between a FRET-pair (inspired by Alex M. Mooney).
Mass Spectrometry
To estimate the product yield of catalyzed reactions by Sortase A7M we performed mass spectrometry. The tested molecules can be distinguished between products and educts due to desorption and ionization. Therefore, we used the electrospray ionization (ESI) technique for the mass spectrometry. This technique has a low resolution but is a very soft ionization method, which makes it an optimal method for biological molecules.
Enzyme-linked Immunosorbent Assay (ELISA)
The enzyme-linked immunosorbent assay (ELISA) is an analytical assay frequently utilized for immobilization and verification of different macro molecules. Immobilizing the recognition tag of the sortase on a surface allows us to verify the coupling efficiency of Sortase A7M under certain conditions. Firstly, we functionalized paper to presenting a poly-G peptide sequence on the surface. Using the Sortase A7M, a ZZ-domain carrying a LPETG amino acid sequence is coupled to the short peptide sequence of GGGßA. The ZZ-domain itself shows high affinity to the human IgG antibody FC-domain and therefore allows the following immobilization of IgG. The secondary antibody is an anti-human IgG, Fab-specific antibody carrying the horseradish peroxidase enzyme (HRP). HRP is capable of converting 4-chloro-1-naphthol to benzo-4-chlorocyclohexandienone using hydrogen peroxide. This color-reaction allows us to draw a conclusion about the previous ZZ-domain’s sortase-mediated coupling efficiency since the turnover of the HRP is directly connected to the ligated ZZ-domains.
Results
Characterization of Sortase A7M (and comparison to BBa_K3187016)
How do we measure if our purified sortases are active?
After purification of the sortases, we first performed SDS-PAGEs to verify that they are pure and monomeric. You can see in Fig. 3 that the purifications were successful. Next, we tested if the purified sortases connect two proteins that carry the important Sortase-recognition tags, N-terminal polyG and C-terminal LPETGG. Therefore, we added the sortases to a mix of GGGG-mCherry and mCherry-LPETGG. The reactions were performed in different buffers, at different enzyme-to-substrate ratios and for different time spans. We performed an SDS-PAGE, and prior to Coomassie staining, we recorded fluorescent images of the gel. Thereby, we could identify mCherry bands in the gel.

Figure 3 : SDS-PAGE of Sortase A7M and Sortase A5M where the bands show up at approximately 15 kDa. Our estimated size for Sortase A7M was 17.85 kDa, and for Sortase A5M 18.07 kDa. This confirms the result shown on the gel, since the band of Sortase A5M is a little higher than the one of Sortase A7M.


Figure 4 :
a) Fluorescence gel of the sortase-reaction of GGGG-mCherry and mCherry-LPETGG
mediated by Sortase A7M incubated for 2 h and
4 h each. Reaction solutions were mixed with different ratios from enzyme to
substrate concentration(1:3;1:10) and each incubated in two different buffers(Tris-HCl
and Ammoniumdicarbonat).
Product bands at a height of about 57 kDa can be seen in lane 4, 5, 6, 8, 9 (from
left to right). The bands below the product at about 38 kDa could be semi-denatured
mCherry dimers.
b) Fluorescence gel on top of the coomassie-stained gel of the sortase-reaction
of GGGG-mCherry and mCherry-LPETGG mediated by Sortase A7M incubated for 2 h
and
4 h each. Reaction solutions were mixed with different ratios from enzyme to
substrate concentration(1:3;1:10) and each incubated in two different buffers (Tris-HCl
and Ammoniumdicarbonat).
Product bands at a height of about 57 kDa can be seen in lane 4, 5, 6, 8, 9 (from
left to right). The bands below the product at about 38 kDa could be semi-denatured
mCherry dimers. Additionally, Sortase A7M can be seen at 17 kDa.The
unprocessed mCherry monomers can be seen at 28 kDa.
As shown in Fig. 4, under certain conditions, a product band appeared at the expected size of 57.3 kDa (28.5+28.8 kDa). From this first activity test, we draw three conclusions:
- Our purified Sortase A7M is active
- The enzyme-substrate ratio affects the product yield
- The duration of the reaction affects the product yield
Additionally, TRIS buffer seems to alter the coomassie staining efficiency of Sortase
A7M.
This endpoint measurement gave us a first impression that our Sortase A7M works nicely.
Of
course, we wanted to further characterize the parameters of the reaction. When we
understand
the Sortase better, modification of our VLPs will become more straightforward.
How do we measure sortase reaction kinetics?
In the above described assays, we noticed the impact of enzyme-substrate ratio and reaction duration on the overall product yield. We thought about how to further measure the kinetics of the sortase reaction. In the literature, sortase reaction kinetics are often measured by FRET-assays. Therefore, we designed a suitable FRET-assay.
Development of a new FRET pair
For characterization of the reaction kinetics of Sortase A7M, Sortase A5M and Sortase A, we decided to develop a suitable FRET pair. In order to find an optimal FRET pair, we first recorded an emission and absorption spectrum of 5-Carboxytetramethylrhodamin-LPETG (TAMRA) and GGGG-mCherry to verify the suitability for the FRET effect, checking for a possible overlap between the donor's emission and the acceptor's excitation.

Figure 5 : Design of a FRET-pair of 5-TAMRA-LPETG (TAMRA) and GGGG-mCherry (mCherry). In this configuration TAMRA acts as donor and mCherry as acceptor. When the two fluorophores are not linked via the substrates of the sortase only TAMRA is being excited. After sortase mediated ligation of the two substrates mCherry is the fluorophore being excited via the FRET and the emission of mCherry intensifies. Meanwhile, the emission of TAMRA decreases.
TAMRA is a chemical fluorophore that has an absorbance maximum at 543 nm and an
emission
maximum at
570 nm. The
terminal carboxy
group of the dye was linked via a lysine linker to the LPETG sequence (see
Fig. 5).
mCherry has
an N-terminal poly-glycine sequence and can therefore be linked to the LPETG motif of TAMRA
via
the
Sortase A. For a sufficient FRET-effect, it is also necessary that the distance between
donor and
acceptor is lower than the Förster radius.
First, we wanted to identify which concentrations are needed for our experiment, then set up
the
reaction
and measured fluorescence intensities. Over time, a decline in the emission of TAMRA can be
observed as
Sortase A7M/A5M is converting more educts to products.

Figure 6 : The graph shows the excitation and emission spectra of TAMRA and mCherry. Due to the large overlap of TAMRA emission and mCherry excitation it is possible to perform a FRET with this pair of fluorophores. The graph show the relative fluorescence unit (RFU[%]) in relation to the excited/emitted wavelength [nm]. The peaks are normalized to 100 %.
The emission and excitation spectra of TAMRA and mCherry exhibit an overlap of emission of TAMRA and excitation of mCherry. Based on this output, a FRET-assay for the kinetics of Sortase A7M was performed to confirm whether the FRET-pair is working. As TAMRA is excited with light of a lower wavelength than mCherry, the former serves as FRET donor and the latter as acceptor. We chose the excitation wavelength at 485 nm to prevent unnecessary “leak” excitation of mCherry. Nevertheless, an excitation of mCherry could not be excluded and may have negative effects on the visibility of the FRET.

Figure 7 : Spectrum of the negative control of TAMRA and mCherry, without Sortase A7M, over the course of 20 min in 5 min intervals. Depicted are the emission wavelengths against the RFU.

Figure 8 : Spectrum of TAMRA and mCherry, with Sortase A7M, over the course of 20 min in 5 min intervals. Depicted are the emission wavelengths against the RFU. The sortase-mediated ligation results in a decline of both emission peaks.
The analysis of the data shown in Fig. 7 confirmed the aforementioned suspicion that mCherry is also excited at 485 nm, which makes differentiation of the fluorescence more difficult. Furthermore, Fig. 8 shows that the difference in the decline of TAMRA is not significant. Accordingly, a decline in the emission maximum of TAMRA over time is also visible in the negative control. One reason might be bleaching of TAMRA through the excitation by the laser. Nevertheless, conversion by the Sortase A7M can be observed by comparing the results with the negative control.

Figure 9 : Sortase reaction in TAMRA-mCherry-FRET after subtracting the negative control. Depicted is the difference in RFU over time [min]. Within the first 20 min of the substrate conversion is the quickest. At 30 min a plateau is reached. After 60 min starts catalyzing the reverse reaction. The mean ΔRFU value was normalized to zero for better visualization. The ΔRFU refers to the difference between the negative control without the respective sortase at 570 nm.
To confirm the functionality of the Sortase A7M, another more sufficient FRET-pair was developed. The measured absorbance and emission spectra indicated that TAMRA and superfolder green fluorescence protein (sfGFP) are a possible FRET-pair.

Figure 10 : The graph shows the excitation and emission spectra of TAMRA and mCherry. Due to the large overlap of sfGFP emission and TAMRA excitation it is possible to perform a FRET with this pair of fluorophores. The graph show the relative fluorescence unit (RFU[%]) in relation to the excited/emitted wavelength [nm]. The peaks are normalized to 100 %.

Figure 11 : Design of a FRET-pair of 5-TAMRA-LPETG (TAMRA) and GGGG-sfGFP (sfGFP). In this configuration sfGFP acts as donor and TAMRA as acceptor. When the two fluorophores are not linked only sfGFP is being excited. After sortase-mediated ligation of the two substrates, TAMRA is the fluorophore being excited via FRET and the emission of TAMRA intensifies. Meanwhile, the emission of sfGFP decreases.
The transfer of energy from sfGFP to TAMRA can be seen by the decrease in emission of sfGFP and increase in emission from TAMRA. Compared to TAMRA as an acceptor, the sfGFP bleaches significantly less and is consequently more suitable as a donor for FRET. Furthermore, the afore mentioned problem of simultaneous donor and acceptor excitation seems to be solved. It seems that we have found a FRET-pair with superior properties.

Figure 12 : Spectrum of the negative control of TAMRA and sfGFP, without Sortase A7M, over the course of 25 min in 5 min intervals. Depicted are the emission wavelengths against the RFU.

Figure 13 : Spectrum of TAMRA and sfGFP, with Sortase A7M, over the course of 25 min in 5 min intervals. Depicted are the emission wavelengths against the RFU. The sortase-mediated ligation results in a decline of both emission peaks.
Due to the collected data of both FRET-pairs we decided to use the TAMRA-LPETG and GGGG-sfGFP FRET-pair for further characterization of our Sortase A variants. Two reasons justify this decision:
- TAMRA bleaches stronger than sfGFP when excited with a laser.
- The spectral overlap between TAMRA and mCherry disturbs “clean” energy transfer, thus the FRET-effect would be less visible and could not be used for analysis of the sortase-mediated reaction.
For recording of sortase reaction parameters we recommend using the FRET-pair sfGFP-TAMRA. As this pair of fluorophores proved to have near perfectly aligned spectra and since the bleaching effect is visibly lower on sfGFP than on TAMRA, we chose to use this FRET-pair in most of our following assay. Nevertheless, we do not rule out the use of TAMRA-mCherry as a FRET-pair since we used it in several FRET-assays as well.
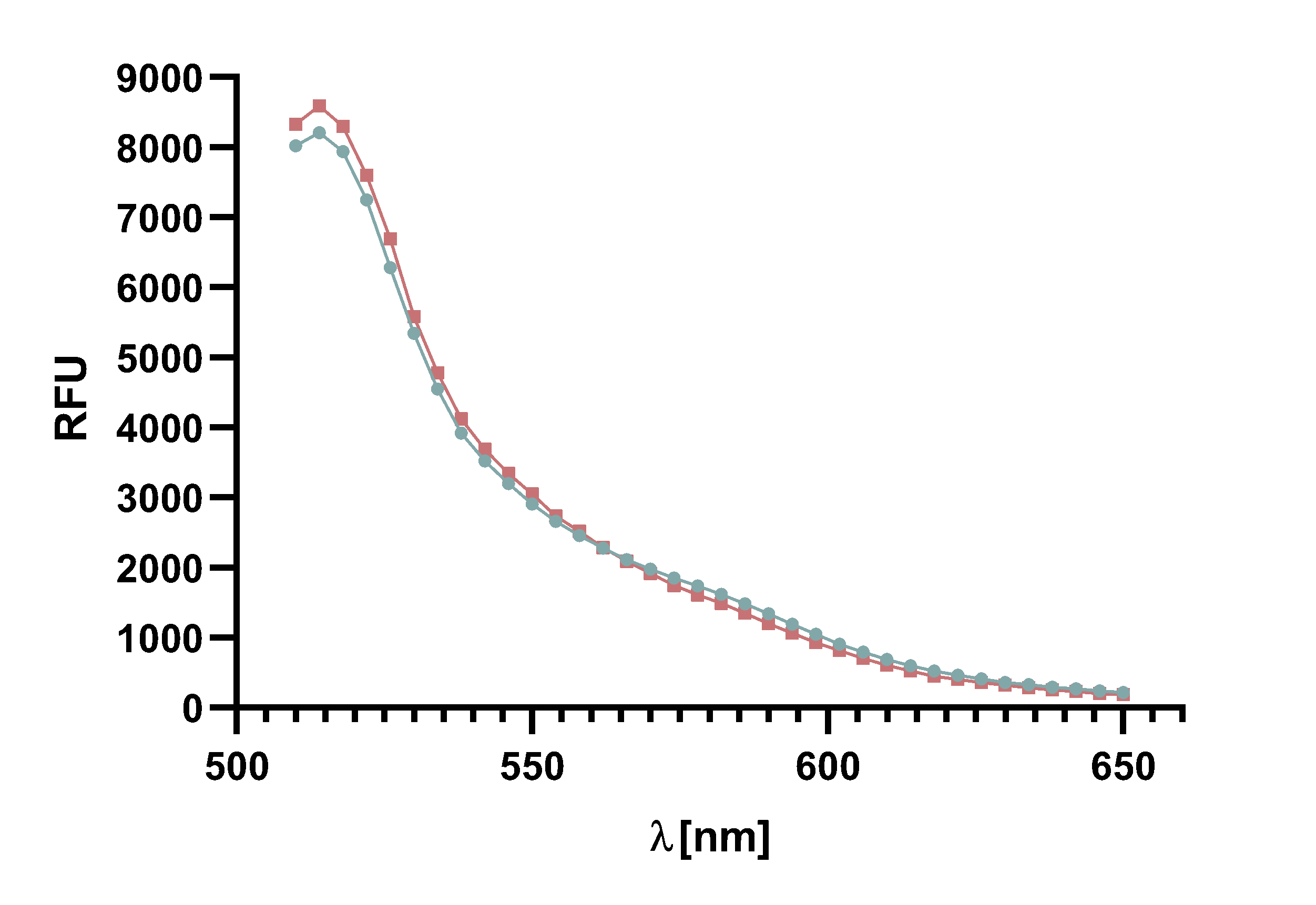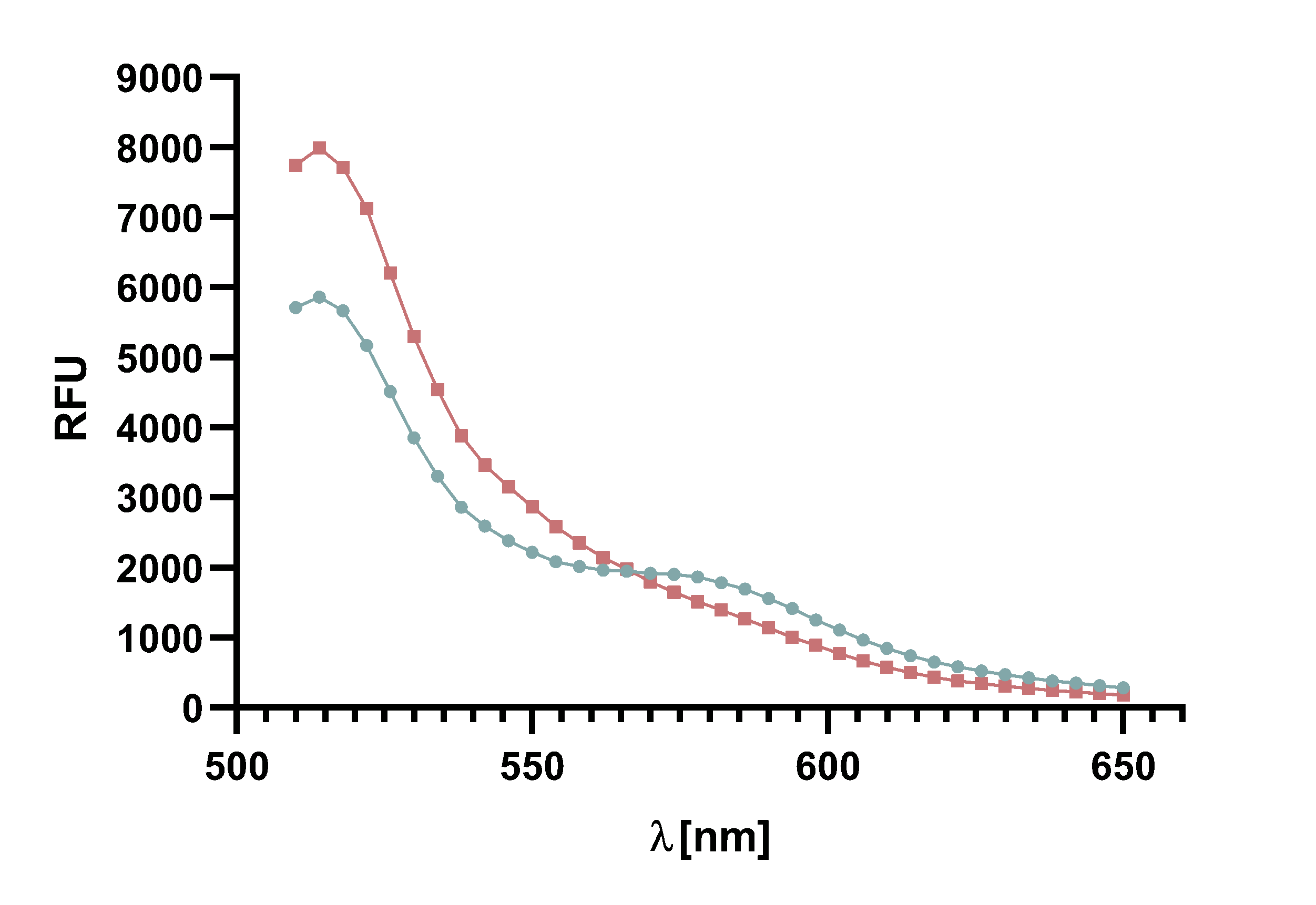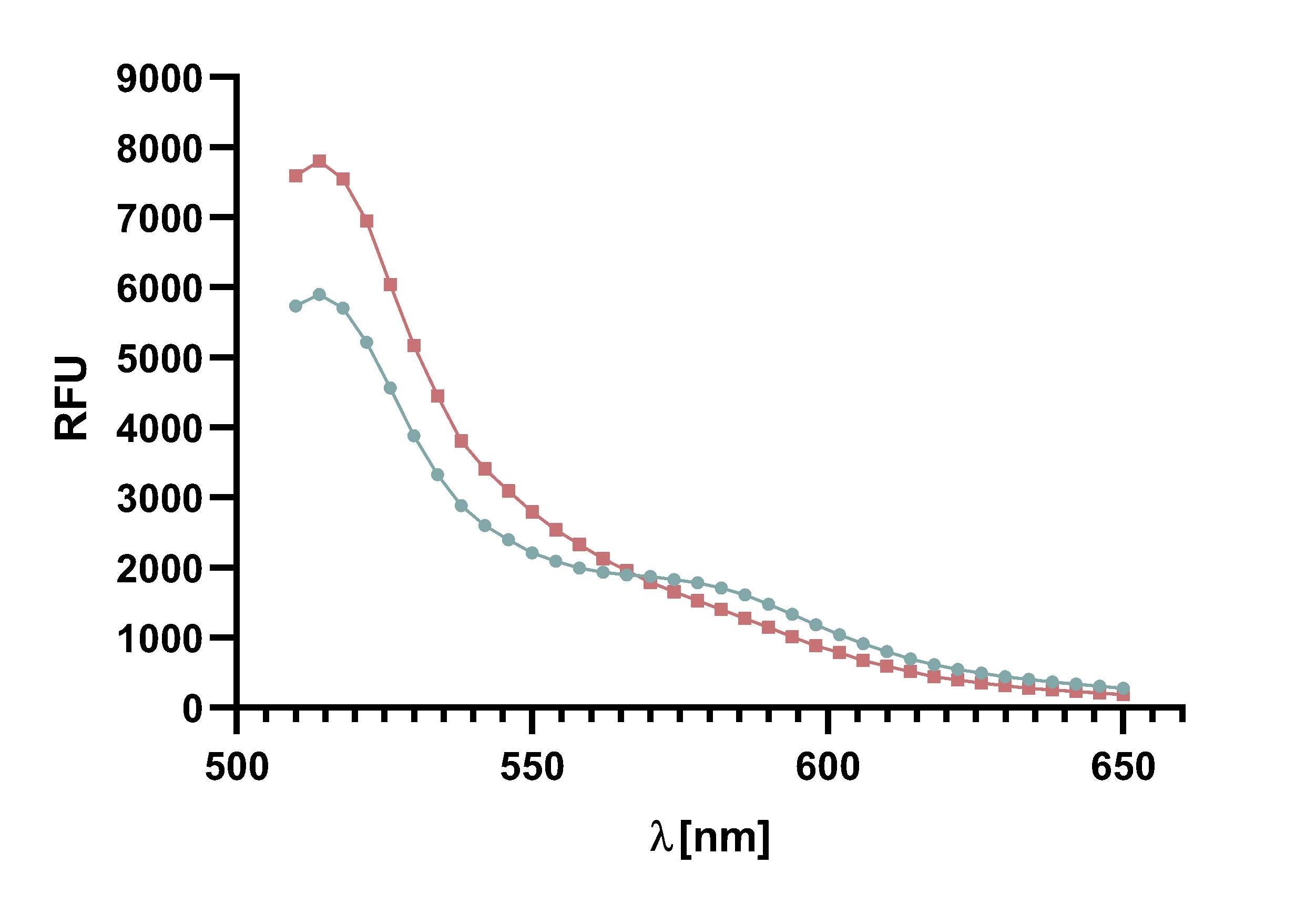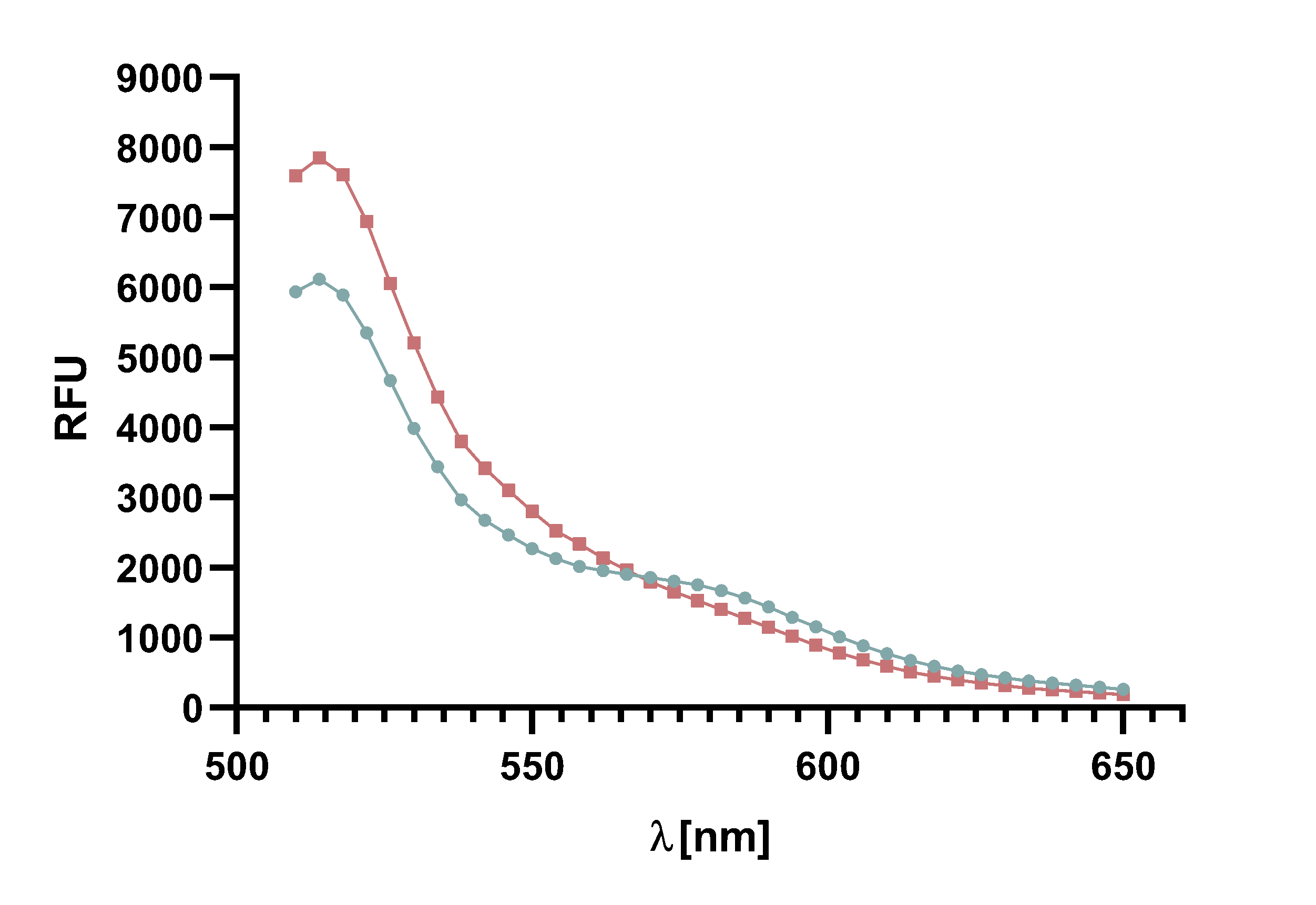
Figure 14 : Animation of Sortase A7M enzyme kinetics over the course of 3 h. The reaction speed increases radically in the beginning moving from RFU 8000 to RFU 6000 at λ = 550 nm where a plateau is reached (blue). The negative control (orange) is also reduced in its RFU due to bleaching. Nevertheless, a peak at λ = 580 nm arises already after short reaction time. This peak indicates the successful Fluorescence Resonance Energy Transfer.
Why are enzyme-substrate ratio and duration important parameters of the sortase reaction?
In one of our first FRET experiments, we addressed the simple theory: More sortase in the reaction mix improves the initial product formation. For this, we used the TAMRA-LPETG : GGGG-mCherry FRET pair. We measured the FRET change over time in a multiwell platereader (Fig. 15).

Figure 15 : Reaction kinetics of Sortase A7M in different concentrations at same level of substrate concentration (6 µM TAMRA-LPETG and (M)GGGG-mCherry). The light blue graph shows the reaction when the sortase concentration is higher (5 µM) and lower concentration (1 µM), in the dark blue graph. Wen the enzyme concentration is lower the maximum in substrate conversion is reached later. The light blue graph also shows a slight decline in product concentration from 60 min onwards.The ΔRFU refers to the respective negative control without each sortase at 570 nm. The mean ΔRFU value was normalized to zero for better visualization.
However, in this assay we observed a striking feature of the sortase reaction. In the reaction with more Sortase A7M present, the FRET change started to decrease after a certain maximum was reached! We suspected some kind of dead-end product formation, as the sortase does also catalyze the reverse reaction of product to educts. Therefore, the overall reaction duration is a very important parameter. We gathered more details about the role of the reverse reaction during our comparison of Sortase A7M and Sortase A5M. Just keep reading if you want to know more!
Who wins - Sortase A7M or Sortase A5M
In our introduction we described that Sortase A7M and Sortase A5M are both fascinating enzymes, although each of them has a unique „selling point“. Sortase A5M is faster, whereas Sortase A7M is Ca2+-independent. We confirmed both of these points in extensive FRET-assays. According to the literature, Sortase A5M works best with a Ca2+-concentration of 2 mM. In contrast, Sortase A7M is a calcium-independent mutant of the enzyme. Moreover, Ca2+ even seems to inhibit this enzyme variant slightly .
Firstly, we confirmed that in contrast to Sortase A5M, Sortase A7M is Ca2+-independent. The results are shown in Fig. 16 Sortase A7M also works in presence of Ca2+, but these FRET experiments made us suspect that Ca2+ may even inhibit Sortase A7M.

Figure 16 : Sortase A7M FRET-assay of connecting TAMRA-LPETG with GGGG-sfGFP with and without Ca2+. The Sortase A7M reaction was measured with 6 mM Ca2+ every minute. Sortase A7M reaction without Ca2+ was measured every three minutes. It is shown that this enzyme variant works with calcium and without calcium as well, although it seems like Sortase A7M is slightly inhibited due to the presence of calcium which explains why the left graph is lower than the right one. The ΔRFU refers to the respective negative control without each sortase at 514 nm. The mean ΔRFU value of the duplicates was normalized to zero for better visualization.
According to the results of this assay, Sortase A7M is definitely Ca2+-independent, since it shows linking activity without calcium in the vicinity. The enzyme mutant also works in presence of Ca2+, but these FRET experiments made us suspect that Ca2+ may even inhibit Sortase A7M, since it shows less activity with calcium around than without calcium.
To better address this question, an ELISA was performed. Therefore, a piece of paper functionalized with GGGβA was connected to a protein domain, which binds antibodies to the LPTEG-tag. The results are shown in Fig. 18.

Figure 18 :
Absorbance at 450 nm at a temperature of 23.8˚ C
In well 1 additional 10 mM Ca2+ were
added which was not the case in well 2. Well 3 serves as a negative control since
the enzyme is missing in this reaction
As shown in Fig. 18, the highest absorption was measured in well 2. Thus, Sortase A7M works more efficiently when no Ca2+ is around. The absorption is also relatively high for the negative control, which can be explained by poor washing before the substrate for Horeseradish peroxidase (HPR) was added. This assay shows the functionality of Sortase A7M even in context of surfaces since we confirmed that Sortase A7M is able to connect tags attached to paper. This shows that the surface structure is not a relevant factor for the enzyme.

Figure 19 : Comparison of the reaction speed of Sortase A5M with Ca2+ and Sortase A7M without Ca2+, therefore each working under optimal conditions. The kinetics were measured via a FRET connecting TAMRA-LPETG and GGGG-sfGFP. The ΔRFU refers to the respective negative control without each sortase at 514 nm. The mean ΔRFU value of the triplicates was normalized to zero for better visualization.
When we compare the reaction speed of Sortase A5M and Sortase A7M, Sortase A5M is the clear winner (see Fig. : 19). However, this means of course that the reverse reaction is also faster in the case of Sortase A5M. Consequently, Sortase A7M is the best variant for in vivo modification of our VLPs as it is Ca2+-independent. On the other hand, Sortase A5M is a suitable enzyme variant for in vitro modification due to its high efficiency.
What about other substrates?
Primary Amines
The literature
describes Sortase A7M as somewhat „promiscuous“ towards other substrates than
GGGG(polyG) as long as the substrate possesses a primary amine. The
Sortase A7M used for this assay was stored in the fridge at 4 °C for two
weeks.
The substrates were TAMRA with a KLPETG bound to TAMRA via the lysine side chain and
3-azidopropanamine as the example for a primary amine. The reaction was performed for
two
hours at 37 °C.
It was then analyzed by electron spray ionization mass spectrometry
(ESI-MS) (Fig. 20).

Figure 20 : Mass spectrum before the reaction of TAMRA-LPETG with 3-azidopropanamine showing the educt at 1054 g/mol.
Fig. 20 shows the educt-peak in the mass spectrum. TAMRA with the LPETG-tag weighs 1054 g/mol. Shown above in green is the single charged molecule at 1054.27 g/mol and the double charged molecule at 528.75 g/mol.

Figure 21 : Mass spectrum after the reaction of TAMRA-LPETG with 3-azidopropanamine showing the product at 1079.37g/mol.
Furthermore, we can conclude that the Sortase A7M accepts any primary amine as a substrate. However, mass spectrum does not show the ratio of educt and product, which is why we cannot estimate whether the turnover is as high as when using a polyG-tag as substrate. Additionally this assay confirms our suspicion that the Sortase A7M is stable at 4 °C and still functional if stored at said temperature for at least two weeks.
Yield
For the characterization of Sortase A7M an assay was designed to show the coupling efficiency between the TAMRA-LEPTG and the tetrapeptide GGG-Beta-Alanin (GGGβA) catalyzed by the Sortase. The Sortase reaction was performed for 1h at 30˚C and was stopped by enzyme separation through centrifugal filtration. For analysis mass spectrometry (ESI-MS) was used. The mass spectrometry enables differentiation between products and educts. It allowed us to make an estimate of the product yield. The calculated theoretical molecular masses are 1054 g/mol for TAMRA and 1240 g/mol for TAMRA-LPETGGGβA. Therefore, peaks are expected at mass/n, with n ∈ N. By comparison of the number of corresponding peaks, estimation of the product yield is possible as both molecules possess the same amount of ionizable groups and thus the difference in the ionizability of both molecules is negligible.

Figure 22 : Mass spectrum of the sortase-mediated ligation of TAMRA-LPETG and GGGβA showing the difference in height of the educt-peak and the product-peak which can be used to estimate the yield of our Sortase A7M.
In Fig. 22 the 621.56 peak can be assigned to the TAMRA-LEPTGGGβA and the 528.85 to the TAMRA-LPETG. The count ratios of the two molecules mentioned show an excess of the product.
Is Sortase A7M able to attach cargo to P22 coat protein?
We performed the linking reaction with CP-LPETGG and GGGG-mCherry as substrates and applied them to an SDS-PAGE. We saw products at the expected size (28 kDa + 49 kDa = 77 kDa) thus the requirement is fulfilled. However, a lot of additional bands appeared that we did not expect. These bands also appeared when only Sortase A7M and CP were mixed.


Figure 23:
a) Sortase A7M band is at expected height (17.85 kDa). The two negative controls containing only GGGG-mCherry (28 kDa) and CP-LPETGG (49 kDa) at the expected respective heights. b) Shown are sfGFP-SP and CP-LPETGG each incubated with both Sortase A7M and Sortase A5M. Both gels display multimers when coat and a sortase variant are in a sample together.
To investigate this issue, we had a look at the literature and found a matching description in the publication of Patterson et al.. They performed a similar experiment with P22 capsid proteins and observed the same multimers in their SDS-PAGEs . Comparing both SDS-PAGEs, we came to the following assumption:
Because of the promiscuity of Sortase A7M to accept primary amines as substrates, as we discussed previously, the formation of CP multimers occurs, unspecifically catalyzed by Sortase A7M.
Parallel to these experiments, we successfully modified the exterior of pre-assembled VLPs in vitro (VLP assembly). These modified VLPs were homogenous and overall correctly assembled. Therefore, we conclude that the described multimer problem only occurs when Sortase A7M encounters free CP.
Does methionine affect Sortase linking?
Sortase A7M preferably attaches N-terminal poly-G to C-terminal LPETGG. However, the first amino acid of a protein is methionine (to be specific, formylmethionine in bacteria). For our constructs that possess N-terminal polyG-tags, we have to ask ourselves the question: If the initial methionines are not cleaved off after the proteins have been produced, will this interfere with the Sortase reaction?
To investigate this, we cloned and purified two other proteins: TVMVsite-GGGG-mCherry and TEVsite-GGGG-sfGFP. Then we treated these proteins with the respective proteases, resulting in *GGGG-mCherry and *GGGG-sfGFP. Following this *GGGG-mCherry was then compared to (M)GGGG-mCherry which we used in all previous assays. Assays were also conducted on Fig. 24 the processed *GGGG-sfGFP substrate. Fig. 24 confirmed our assumptions that the unprocessed substrate cannot be linked to the sorting motif via Sortase A7M. Subsequently, *GGGG-sfGFP (after protease digest) demonstrate successful linkage via sortase-mediated ligation.
Due to these findings we modified our VLPs with *GGGG-sfGFP.

Figure 24 : Sortase-mediated ligation of TAMRA-LPETG and GGGG-sfGFP (with TEV cleavage site) one cut with TEV protease and one not. The sample with the unprocessed substrate shows no increase in RFU. In contrast the processed substrate shows a clear increase in ΔRFU. After 90 min the reverse reaction begins.The ΔRFU refers to the respective negative control without each sortase at 514 nm. The mean ΔRFU value of the duplicates was normalized to zero for better visualization.
We performed FRET-assays with TAMRA-LPETG and either of the following reaction partners:
- (M)GGGG-mCherry, a protein sample that might still carry an N-terminal methionine
- *GGGG-mCherry that does not carry any additional N-terminal residue
Before the FRET-assay was started, we adjusted the mCherry-concentrations of both fluorescent protein solutions to the same level. To do so, we diluted them until both showed the same fluorescence at 610 nm.


Figure 25: FRET of the sortase reaction connecting TAMRA-LPETG and GGGG-sfGFP mediated by Sortase A7M. The concentration of the Sortase A7M and TAMRA-LPETG was kept at the same level why the concentration of sfGFP was either 10,7 µM or 1,4 µM. The graphs show that the reverse reaction happens earlier if the GGGG-substrate concentration is lower. The ΔRFU refers to the respective negative control without each sortase at 514 nm. The mean ΔRFU value of the duplicates was normalized to zero for better visualization.
Strikingly, only the (M)GGGG-mCherry construct showed a clear decrease in delta RFU after the maximum delta RFU was reached (at about 160 min).

Figure 26 : Sortase-mediated ligation of TAMRA-LPETG and GGGG-mCherry one cut with TVMV protease and one with a methionine infront of the GGGG-tag. As visible the reverse reaction happens earlier if the methionine is not cleaved of the GGGG-tag. The ΔRFU is referring to the negative controls without Sortase A7M at 570 nm. The mean ΔRFU value of the tripicates was normalized to zero for better visualization.
We assume the following: Although we adjusted the overall mCherry concentration by fluorescence, we cannot determine the absolute amount of MGGGG-mCherry in the (M)GGGG-mCherry sample. However, if this amount was relatively high, the effective substrate concentration that could enter the sortase reaction would be low. That is because MGGGG is a worse sortase substrate than GGGG – if any at all. If we furthermore consider that a low substrate concentration correlates with a faster reverse reaction, we can explain the observed decrease in delta RFU for the (M)GGGG-mCherry sample that contrasts the delta RFU trend of the *GGGG-mCherry sample.
On this basis we can assume that a certain, yet unknown portion of the (M)GGGG-mCherry sample still carries an N-terminal methionine.
These FRET-assays let us assume that methionine disturbs or at least interferes with the sortase reaction mechanism. Indeed, our modeling suggests that methionine affects the interaction of polyG and the flexible loop near the active site of Sortase A7M. Click here if you want to know more about our modeling results!
This strengthens our hypothesis: If there is any amino acid in front of the poly-glycine sequence, substrate binding to Sortase A7M is negatively influenced.
Modeling
Introduction
In synthetic biology, theoretical models are often used to gain insights, predict
and
improve
experiments. In our project we are modifying Virus-like particles (VLPs) by
attaching
proteins to the
surface of the P22 capsid
through a linker. The linking is
catalyzed using
the enzyme Sortase A7M, which is a calcium-independent mutant of the wild type
Sortase A
from Staphylococcus aureus. We performed
modeling to predict the unknown structure of the
Sortase A7M, to improve the linker between proteins and therefore optimizing the
modification
efficiency.
Here Rosetta Comparative Modeling was performed to predict the structure of
Sortase A7M. The generated structure was characterized in terms of the molecular
mechanisms and binding affinities of ligand peptides using molecular dynamics
and docking simulations.
Structure determination
In silico modeling and simulation of proteins requires a 3D structure, which can be obtained from the RCSB Protein Data Bank. However, if no 3D structures are annotated, as it is the case with sortase A7M, the structure has to be determined by other means. The structure prediction of sortase A7M was done using two different approaches.
RosettaCM
Results
The simulation of our Sortase A7M yielded 15,000 structures which have been compared using the Rosetta scoring functions (talaris2013). From the 15,000 structures generated, we inspected the ten best scoring structures.
As can be seen in Fig. 27, the most prominent differences can be found in the regions close to the N- and C-terminus. As fluctuations in those regions are not untypical, we decided to use the best scoring structure, candidate S_14771 (Fig. 28), as the input for the simulations to follow.

Figure 27 : The structural alignment of the ten best scoring sortase structures displaying minor differences with the exception of the C- and N-terminal regions. N- and C-terminal regions tend to show strong fluctuations, thus it is unsurprising to find the terminal regions to be unaligned

Figure 28 : Sortase A7M candidate S_14771 created through RosettaCM.
In order to evaluate the secondary structure of the Sortase A7M candidate S_14771 a Ramachandran plot has been created and compared to the five sortases used as input for the comparitive modeling.



Figure 29: The Ramachandran plot of randomly sampled proteins and the input structures of the comparative modeling show similar secondary structures. Secondary structure analysis of both sortase candidates reveals absence of secondary structures for the ML candidate. This is not the case with candidate S_14771 as the Ramachandran plot shows all relevant structures.
The Ramachandran plot (Fig. 29) showing α-helices and β-sheets is a strong indicator of a successful structure determination, as those secondary structures are crucial for the functionality of sortases.
Conclusion
We used monte-carlo simulations
to determine the structure of the mutated transpeptidase Sortase A7M.
As a first investigation of the secondary structure a Ramachandran plot of
candidate S_14771 was made. The plot showing secondary structures was a first
indicator
of successful structure prediction.
The approach, using Rosetta Comparative Modeling, yielded
15,000
structures scored with the talaris2013 scoring function. The ten
best structures
were aligned and exhibited almost identical secondary structures
(Fig. 27).
The greatest structural differences are present in the N- and
C-terminal regions. Since terminal regions tend to fluctuate more strongly than
non-terminal segments of the protein, we deemed those fluctuations
non-relevant for the proteins functionality.
Being the best scoring candidate, structure S_14771 was analyzed
structurally using a Ramachandran plot (Fig. 29). The plot shows all the
relevant and typical structures sortases exhibit and serves as an indicator for
a successful structure prediction.
In the steps to follow, a molecular dynamics (MD)
simulation was performed on both structures.
Molecular dynamics
Results
The first possible indicators of a stable protein structure are converging root-mean-square deviation (RMSD), small root-mean-square fluctuation (RMSF) values as well as converging radii of gyration. Using the Python software package and the module Biotite we calculated these quantities and plotted the result for candidate S_14771.

Figure 30 : The RMSD is one of three main indicators of a stable protein structure of the MD simulation of S_14771 over the period of 200,000 ps. As time progressed the RMSD increased with a smaller slope. The value stabilizes at a time of 110,000 ps and fluctuated around the value of 6 Å.

Figure 31 : The prominent fluctuations of the residues from ranges 105 to 115 might indicate a binding site or another form of functional structure. The radius of gyration, just as the RMSD Fig. 30, stabilizes around a simulation time of of 110,000 ps and converges towards a value of 16.7 Å.

Figure 32 : The fluctuations (RMSF) of most residues appear insignificant compared to the first, the last residues and the residues close to residue 110 . Typically the N- and C-terminus tend to fluctuate more intensively due to the lack of stabilizing structures. The prominent fluctuations in the range of residue 105 to 115 can indicate a binding site or another form of functional structure.
Typical RMSDs and radii of gyration converge towards a value dependent on the size of the protein. Convergence of those quantities can be interpreted as a stable state of the protein structure. As it can be seen in Fig. 30 and Fig. 31 both the RMSD and the radius of gyration stabilize at the same time as the simulation reaches 110,000 ps (110 ns), suggesting a now stabilized structure of candidate S_14771 solvated in water. Another indicator of a functional protein is the RMSF. Instead of being averaged over all atoms, the RMSF is averaged over time with respect to each amino acid. It provides insights in both protein stability and functionality. Fig Fig. 32 reveals the RMSF of residues 105 to 115 to be significantly higher than that of other residues. This hints at the presence of a functional unit along these residues. As commented on in the section describing our structure prediction approaches, the N- and C-terminal regions tend to fluctuate more strongly as a result of the absence of stabilizing structures.
To further analyse the potential binding site, we performed a Principle Component Analysis.
Principle component analysis
To analyze our system further Principle Component Analysis (PCA) was performed using GROMACS. By applying PCA to a protein it is possible to gain insights into the relevant vibrational motions and thereby the physical mechanism of the protein.

Animation 33: A Principle Component Analysis of a fast (blue) and a slow (red) mode showing the most prominent movements of the Cα-chain of candidate S_14771. Both modes show movement of the β6/β7 loop consisting of residues 105 to 115 towards the active site . Thus we can assume that the closing β6/β7 loop is involved in the reaction mechanism.
The results from the Principle Component Analysis of candidate S_14771 (Amination 33) show a movement of the residues 105 to 115 towards the active site, supporting our theory that residues 105 to 115 are important for the reaction mechanism. Since the most relevant eigenvector (red), which shows the most relevant movement of the sortase, moves further towards the active site, it is possible that the β6/β7 loop either closes the binding site of the ligand peptides or even transports one peptide towards the other.
Conclusion
Candidate S_14771 that was generated using RosettaCM appears to be a fitting candidate not only due to successful analyses, but also due to the residues of the active site being in close proximity. Not the RMSF but also Principle Component Analysis indicate that residues 105 to 115 forming the β6/β7 loop are crucial for the molecular mechanism of our model.
Docking
Now that the binding site of the Sortase had been found, the peptide ligand needed to be inserted into the binding site to create a peptide-protein complex. The procedure of choice for the introduction of a ligand into the binding site of a protein is called docking. In the following sections, we will present the protocol and methods we used as well as the results they yielded.
Results
For sequences MGGGGPPPPPP(M-polyG), GGGGPPPPPP(polyG) and PPPPPPLPETGG(LPETGG) 50,000 structures have been created and clustered. After the clustering the sample consisted of 100 structures of docked complexes.

Figure 34 : The three best scoring structures (total score, interface score, reweighted score) of the LPETGG-tag are shown. Only two results are visible as the best reweighted score candidate is identical to the best interface score candidate. The reacting section of the LPETGG-tag namely glycine is colored yellow as is the active site. The glycin of both ligand peptides is facing the active site.
Analysis of the scores has shown a similar score for all the three dockings. The best scoring results of the LPETGG docking show a tendency of the glycines to face the active site while also being in close proximity to the active site.

Figure 35: The three best scoring structures (total score, interface score, reweighted score) of the polyG peptide are shown. Only two results are visible as the best reweighted score candidate is identical to the best interface score candidate. Instead of facing the active site (yellow) the reacting glycines (yellow) appear to interact with the β6/β7 loop of the sortase.

Figure 36: The three best scoring structures (total score, interface score, reweighted score) of the poly-g peptide are shown. Only two results are visible as the best reweighted score candidate is identical to the best interface score candidate. Concerning the M-poly-G peptide no uniform directional orientation can be observed. The structure with the best interface score (light blue) is oriendted towards the loop while the structure with the best total/reweighted (dark blue) is oriented towards the β-sheets.
Fig. 34 shows the docking result of the LPETGG peptide to the sortase. The results shown are the best scoring structures of the clustering with respect to the total score, interface score and reweighted score. As the best scoring structure is the same for the total score and the reweighted score only two peptides are shown. This also applies to Fig. 35 and Fig. 36. For both results the reacting glycin residues (yellow) are facing the active site. Additionally, the same residues are in close proximity to the active site.
The figures Fig. 35 and Fig. 36 show the docking of the both polyG and M-polyG. While polyG results align well and seem to be interacting with the β6/β7 loop rather than with the active site, this does not seem to be the case for M-polyG. Instead of both structures interacting with the β6/β7 loop or active site one (best interaction score; dark blue) interacts with the β6/β7 loop and the other (best reweighted/total score; light blue-gray) appears to interact with the active site.

Figure 37: The close up of the M-polyG peptide (best total/reweighted score) indicates an interaction of methionine with arginine139 and cysteine126.

Figure 38: Methionine of the result with the best interface score interacted with the β6/β7 loop rather than the active site. Still the reactive glycine residues appear to be bound to the β6/β7 loop.
As can be seen in Fig. 37 visualizing the result of the the docking simulation (total/reweighted score) suggests an interaction of methionine and two of the active sites namely arginine139 and cysteine126. Fig. 38 shows the interaction of M-polyG with the β6/β7 loop. The glycines still interact with the β6/β7 loop. Instead of binding above the β6/β7 loop, which is the case for polyG as illustrated in Fig. 36, the interaction seems to be influenced by methionine. By interacting with the residues in the β-helix methionine could potentially hinder binding of glycine to the β6/β7 loop by partial immobilization of the peptide. Overall peptide binding and orientation is less uniform compared polyG without the leading methionine, which could be an indicator of lesser binding affinity of M-PolyG towards the β6/β7 loop.
Conclusion
To computationally investigate binding affinities of the polyG and M-polyG as well as the LPETGG tags we performed docking simulations using the Rosetta FlexPepDock application. We used a modified version of the recommended protocol as the modified version was easier to automate and served our purpose better than the standard protocol. From the calculated scores only, we could not see a difference in binding affinities. Thus, we inspected the best scoring structures regarding the total score, the interface score and the reweighted score using PyMOL. Since the best structures with respect to total score and reweighted score were the same for all simulations, only two structures have been inspected per run. A polyproline tag was appended to all the peptides to simulate the modification of the VLPs with a small peptide.
As expected, the results showed that for LPETGG, the glycines of both peptides oriented towards the active site. This is unsurprising as peptides with the sequence LPXTGG are known to be substrate of the Sortase. It was more surprising to see the polyG tag oriented away from the active site since polyG also is a known substrate of the sortase. Both polyG peptides were facing the β6/β7 loop (residues 105 to 115) uniformly and appeared to be interacting with it. The M-polyG peptides did not show a uniform orientation or interaction scheme. On one hand the visualization of the best result concerning the total and reweighted score has shown interaction of methionine with the cysteine126 and arginine139, two residues of the active site. On the other hand, the visualization of the best result with respect to the interface score shows the M-polyG facing the mobile β6/β7 loop. In contrast to the polyG peptide the lacking the methionine, the M-polyG peptide is pulled down below the β6/β7 loop by the methionine interacting with one of the β-sheets leading to the active site. This is not the case with the polgG results, which lie aligned in one plane with the β6/β7 loop.
Modeling Conclusion
For our project it was key to understand and characterize Sortase A7M. As there is no annotated 3D structure for this specific Sortase, an in silico structure determination was performed. Comparative modeling with Rosetta produced valid structures. We used the best structure, candidate S_14771, for extensive characterization. We evaluated the model with regard to its secondary structure using Ramachandran plots. The Ramachandran plot suggested plausible secondary structures.
Molecular Dynamics simulations were used to investigate stability and dynamic properties of the candidate. The RMSD and radius of gyration stabilized over the course of the simulation, a first indicator of an equilibrated structure. Interestingly, RMSF analysis showed strond fluctuations of residues 105 to 115. We further investigated this by performing Principle Component Analysis. Doing so, we extracted the principle movements of the model. We could observe movement of the β6/β7 loop towards the active site, suggesing the presence of a binding site. Consequently, we performed docking simulations.
FlexPepDock was used to conduct the docking simulations with target peptides. Each run yielded 50,000 structures. In multiple steps we reduced the amount of complexes to 100 clusters with respect to total, reweighted and interface score. We extracted the best scoring complexes and investigated interactions.
For LPETGG we observed a uniform binding to the active site, fullfilling our expectation. Strikingly, polyG appeared to bind to the β6/β7 loop in a uniform manner. As it is know from literature polyG is a functioning ligand of sortase. Supported by literature and our data, we postulate the following mechanism: the β6/β7 loop transports bound polyG towards the active site of Sortase A7M, thereby lowering the activation energy of the linking reaction.
As the theory is neither backed up by nor contradicts experimental data, further research is required.
References
For our references, please have a look at our wiki.
Contribution: XHD-Wuhan-Pro-China 2021
literature 1
Bacteria display a variety of proteins on their surface that to enable them to effectively interact with their environment. Gram-positive bacteria use sortase cysteine transpeptidase enzymes to covalently attach proteins to their cell wall, and to assemble pili. Sortases in pathogenic bacteria are frequently important virulence factors, as many of the proteins that they display have key roles in the infection process, such as promoting bacterial adhesion, nutrient acquisition, and the evasion and suppression of the immune response (Cascioferro, Totsika, & Schillaci, 2014; Schneewind & Missiakas, 2012, 2014; Siegel, Liu, & Ton-That, 2016; Spirig, Weiner, & Clubb, 2011). As a result, a significant amount of effort has been put forth to elucidate the mechanism of sortase-mediated catalysis and to discover small-molecule sortase inhibitors that could function as potent antiinfective agents (Bradshaw et al., 2015; Cascioferro et al., 2014; Clancy, Melvin, & McCafferty, 2010; Maresso & Schneewind, 2008; Suree, Jung, & Clubb, 2007). Moreover, sortases have been developed into valuable biochemical reagents to ligate distinct biomolecules together via a covalent peptide bond. This in vitro transpeptidation activity has been harnessed for a variety of useful applications, including among others, covalently attaching proteins to cells, attaching fluorophores or drugs to antibodies, cyclizing proteins, and immobilizing peptides on solid surfaces (Antos, Truttmann, & Ploegh, 2016; Popp & Ploegh, 2011; Ritzefeld, 2014; Schmidt, Toplak, Quaedflieg, & Nuijens, 2017; Schmohl & Schwarzer, 2014; Voloshchuk, Liang, & Liang, 2016).
Sortases perform two distinct functions in bacteria: (1) attach proteins directly to the cell wall or (2) assemble pili, long proteinaceous fibers that project from the microbial surface (Fig. 1). Both reactions are mechanistically related and operate on secreted proteins that contain a C-terminal cell wall sorting signal (CWSS). The CWSS contains a five-residue sortase recognition motif, frequently LPXTG, that is followed by a hydrophobic domain and a positively charged cytoplasmic anchor that retains the protein substrate in the membrane (Schneewind, Model, & Fischetti, 1992). The Sortase A enzyme from Staphylococcus aureus (SaSrtA) has been studied in detail and is archetypal (Mazmanian, Liu, Ton-That, & Schneewind, 1999; Ton-That, Liu, Mazmanian, Faull, & Schneewind, 1999). SaSrtA attaches surface proteins to the cell wall by recognizing an LPXTG motif within the CWSS of its protein substrate (Fig. 2A). Catalysis begins when SaSrtA’s active site cysteine residue nucleophilically attacks the carbonyl carbon in the peptide bond between the Thr and Gly residues in the sorting signal (Fig. 2A, step 1) (Clancy et al., 2010; Connolly & Clubb, 2005). This generates a tetrahedral intermediate that quickly collapses to form a semi-stable thioacyl intermediate in which sortase is covalently linked via its cysteine residue to its protein substrate (Fig. 2A, step 2). SaSrtA then recognizes a second substrate, the cell wall precursor, lipid II (Fig. 2A, step 3) (Perry, Ton-That, Mazmanian, & Schneewind, 2002; Ruzin et al., 2002) and catalyzes a reaction in which the N-terminal primary amine group within the cross-bridge peptide nucleophilically attacks the carbonyl carbon atom within the thioacyl bond. This second transient tetrahedral intermediate resolves into the protein–lipid II product in which the components are joined via a peptide bond (Fig. 2A, step 4) (Perry et al., 2002; Schneewind, Fowler, & Faull, 1995; Ton-That, Faull, & Schneewind, 1997). The lipid II-linked protein is then incorporated into the peptidoglycan via the transpeptidation and glycosylation reactions that synthesize the cell wall. In contrast to attaching proteins to the cell wall, a second type of sortase, frequently called “pilin polymerases,” construct bacterial pili by polymerizing pilin protein subunits (Fig. 2B) (Hendrickx, Budzik, Oh, & Schneewind, 2011; Kline, Dodson, Caparon, & Hultgren, 2010; Mandlik, Swierczynski, Das, & Ton-That, 2008; Siegel et al., 2016; Spirig et al., 2011; Ton-That & Schneewind, 2003). These pilin-assembling enzymes employ a similar transpeptidation reaction as SaSrtA, but instead of using lipid II as a nucleophile to attach proteins to the cell wall, a lysine amino group located within a protein pilin subunit is used as a secondary substrate to attack the sortase–protein thioacyl intermediate (Fig. 2B, steps 3 and 4). This reaction constructs pili by covalently linking protein subunits together via lysine–isopeptide bonds. Both types of sortase-catalyzed processes occur on the extracellular membrane, where the enzyme and its substrate are membrane associated (Cozzi et al., 2011; Spirig et al., 2011; Wu et al., 2012).

Sortase enzymes attach proteins to the cell wall and assemble pili. (A) Overview of anchoring and pilus assembly reactions. A protein that is to be displayed (blue) contains an N-terminal secretion signal and a C-terminal cell wall sorting signal (CWSS). The CWSS contains an LPXTG-like sorting signal sequence that is processed by the sortase, a nonpolar polypeptide segment (black), and a C-terminal segment of positively charged residues (+). After secretion through the Sec translocon, the protein remains embedded in the lipid bilayer via the nonpolar segment within the CWSS. The sortase enzyme then cleaves between the threonine and glycine residues to form a sortase–protein thioacyl intermediate in which the active site cysteine is covalently linked to the carbonyl carbon atom of the threonine. There are two basic types of sortases: (1) cell wall anchoring sortases that attach protein to the crossbridge peptide of the cell wall and (2) pilin polymerase sortases that covalently link pilin subunits together via lysine–isopeptide bonds. In both cases, the enzymes function as transpeptidases. Some sortases are capable of performing both functions, attaching proteins to the cell wall and polymerizing pili.

Mechanism of cell wall protein anchoring and pilus assembly. Sortases perform two basic functions in bacteria: (1) attach proteins to the cell wall and (2) join proteins together to construct pili. (A) In the cell wall anchoring reaction, the sortase and substrate are both membrane bound. The reaction occurs via four distinct steps. Sortase first recognizes a sorting signal motif within the CWSS and nucleophilically attacks the threonine’s carbonyl carbon atom via its active site cysteine residue (for demonstration purposes the LPXTG sorting signal recognized by class-A type enzymes is shown, step 1). The LPXTG sorting signal is then cleaved to produce a sortase–substrate thioacyl intermediate (step 2). Next, the crossbridge peptide from a lipid II molecule nucleophilically attacks the thioacyl intermediate (step 3). Lastly, a new peptide bond is formed between the lipid II molecule and surface protein to produce a protein–lipid II intermediate that is incorporated into cell wall by the transglycosylation and transpeptidation reactions that synthesize the peptidoglycan (step 4). (B) In the pilus assembly reaction, steps 1–2 produce a sortase–substrate thioacyl intermediate, similar to the cell wall anchoring reaction. In this reaction, the sortase recognizes a pilin protein that contains a CWSS. However, a lysine residue within the pilin motif from an adjacent pilin protein performs the nucleophilic attack on the thioacyl intermediate (step 3). A new protein–protein isopeptide bond is formed that covalently links the pilin proteins (step 4). This assembly process is repeated to build an isopeptide-linked pilus shaft that contains multiple pilin proteins. Depending on the type of pilus, distinct tip and base pilin proteins can be located at the ends of the pilus shaft, which are incorporated through a similar mechanism and involve covalent linkages via lysine-derived isopeptide bonds. Finally, the intact pilus is attached to the cell wall via sortase-catalyzed attachment of the pilus to lipid II, similar to cell wall protein display. Some sortases are capable of performing both functions, attaching proteins to the cell wall and functioning as pilin polymerases.
At present, 3330 gene sequences encoding sortase enzymes have been identified within 1098 species of bacteria (Finn et al., 2016). Sortases are primarily found in Gram-positive bacteria, but are also present to a lesser extent in some species of Gram-negative and archaebacteria. Based on their primary sequences, most sortase homologs can be classified into six distinct subfamilies, called class A–F enzymes (Dramsi, Trieu-Cuot, & Bierne, 2005; Spirig et al., 2011). Class A, B, and D enzymes are prevalent in bacteria within the Firmicutes phylum, while class E and F enzymes predominate in Actinobacteria. Class C enzymes are found in both Firmicutes and Actinobacteria. Similar to SaSrtA (a class A enzyme), all sortases contain a His–Cys–Arg catalytic triad (Ilangovan, Ton-That, Iwahara, Schneewind,& Clubb, 2001; Marraffini, Ton-That, Zong, Narayana, & Schneewind, 2004; Ton-That, Mazmanian, Alksne, & Schneewind, 2002), and their primary sequences harbor a highly conserved TLXTC motif that contains the catalytically essential cysteine residue (Clancy et al., 2010). All sortases characterized to date catalyze a transpeptidation reaction that joins an LPXTG-like sorting signal within the CWSS of their protein substrate to an amino nucleophile. However, their sorting signal and nucleophile substrate specificities can vary substantially. These distinct specificities enable microbes to utilize more than one type of sortase to elaborate their surface, with each sortase operating nonredundantly to display or assemble distinct proteins on the cell surface (Comfort & Clubb, 2004; Pallen, Lam, Antonio, & Dunbar, 2001).
A number of excellent reviews have been written that describe the overall function of sortases in bacteria, their development as biochemical reagents, and efforts to discover therapeutically useful sortase inhibitors (Antos et al., 2016; Bradshaw et al., 2015; Cascioferro et al., 2014; Clancy et al., 2010; Maresso & Schneewind, 2008; Popp & Ploegh, 2011; Ritzefeld, 2014; Schmidt et al., 2017; Schmohl & Schwarzer, 2014; Schneewind & Missiakas, 2012, 2014; Siegel et al., 2016; Spirig et al., 2011; Suree et al., 2007; Voloshchuk et al., 2016). In this chapter, we review what is known about their atomic structures and the molecular basis of substrate recognition and catalysis.
Reference
Ge Y, Chen L, Liu S, Zhao J, Zhang H, Chen PR. Enzyme-Mediated Intercellular Proximity Labeling for Detecting Cell-Cell Interactions. J Am Chem Soc. 2019 Feb 6;141(5):1833-1837. doi: 10.1021/jacs.8b10286. Epub 2019 Jan 29. PMID: 30676735.
Literature 2
Strategies for site-specific protein modification are highly desirable for the construction of conjugates containing non-genetically-encoded functional groups. Ideally, these strategies should proceed under mild conditions, and be compatible with a wide range of protein targets and non-natural moieties. The transpeptidation reaction catalyzed by bacterial sortases is a prominent strategy for protein derivatization that possesses these features. Naturally occurring or engineered variants of sortase A from Staphylococcus aureus catalyze a ligation reaction between a five-amino-acid substrate motif (LPXTG) and oligoglycine nucleophiles. By pairing proteins and synthetic peptides that possess these ligation handles, it is possible to install modifications onto the protein N- or C-terminus in site-specific fashion. As described in this unit, the successful implementation of sortase-mediated labeling involves straightforward solid-phase synthesis and molecular biology techniques, and this method is compatible with proteins in solution or on the surface of live cells.
Reference
Antos JM, Ingram J, Fang T, Pishesha N, Truttmann MC, Ploegh HL. Site-Specific Protein Labeling via Sortase-Mediated Transpeptidation. Curr Protoc Protein Sci. 2017 Aug 1;89:15.3.1-15.3.19. doi: 10.1002/cpps.38. PMID: 28762490; PMCID: PMC5810355.