
Difference between revisions of "Part:BBa K3187004"
JonathanFu (Talk | contribs) |
|||
| Line 91: | Line 91: | ||
<h1> Methods</h1> | <h1> Methods</h1> | ||
<h2>Cloning</h2> | <h2>Cloning</h2> | ||
| − | <p>The TEV-site was added to sfGFP through Gibson overhang primers and cloned into the pET24 via <a href="https://static.igem.org/mediawiki/2019/4/44/T--TU_Darmstadt--MethodenFinal.pdf"target="_blank">Gibson Assembly</a>. To verify the cloning, the sequence was controlled by Sanger sequencing by Microsynth Seqlab. | + | <p>The TEV-site was added to sfGFP through Gibson overhang primers and cloned into the pET24 via <a href="https://static.igem.org/mediawiki/2019/4/44/T--TU_Darmstadt--MethodenFinal.pdf"target="_blank">Gibson Assembly</a>. The vector posesses a kanamycin resistance. Expression is controlled by the T7 promoter and induced by IPTG.To verify the cloning, the sequence was controlled by Sanger sequencing by Microsynth Seqlab. |
</p> | </p> | ||
<h2>Purification</h2> | <h2>Purification</h2> | ||
Revision as of 16:33, 21 October 2019
TEV Cleavage Site x GGGG-Tag for Sortase-mediated Ligation X Superfolder Green Fluorescence Protein
Profile
| Name | TEV site-polyG-sfGFP |
| Base pairs | 1028 |
| Molecular weight | 27.8 kDa |
| Parts | T7-Promoter, lac-operator, RBS (g10 leader sequence), TEV protease recognition sequence, polyG-tag, sfGFP, Strep-tag II, T7 terminator |
| Properties | After cleavage by the TEV protease, the polyG tag can be used to fuse sfGFP to the Sortase A recognition sequence(LPTEGG) |
Usage and Biology
The TEV protease is cleaving a protein after a specific sequence between Glutamine and Serine or Glycine
[1]
[2]
.
We are using this to create a free N-terminal polyG sequence in front of sfGFP so we can use it as substrate in a Sortase A mediated reaction
[3]
[4]
[5]
.
sfGFP is a variant of the fluorescence protein GFP that was originally isolated from the jellyfish Aequorea victoria. It has a short maturing time of 13.6 min, has an extinction maximum at 485 nm and an emission maximum at 510 nm.
[6]
[7]
At the end of the sfGFP a strep tag was added to enable easy protein purification.
The part contains a T7 promoter so it can be transcribed by T7 polymerase, and a lac operator so protein expression can be induced by IPTG.
Methods
Cloning
The TEV-site was added to sfGFP through Gibson overhang primers and cloned into the pET24 via Gibson Assembly. The vector posesses a kanamycin resistance. Expression is controlled by the T7 promoter and induced by IPTG.To verify the cloning, the sequence was controlled by Sanger sequencing by Microsynth Seqlab.
Purification
The protein was heterologously expressed in E. coli BL21 and purified with GE Healthcare ÄKTA FPLC. The used affinity tag was Strep-tag II.
SDS-Page and western blot
To verify that the TEV-site with sfGFP was produced, a SDS-PAGE followed by a western blot was performed.
Results
Cloning and Expression
The successful cloning was proven with Sanger sequencing and production with a western blot.
Figure 1: Western blot of all produced and purified proteins.
Fig. 1 shows that sfGFP with TEV cleavage site has a molecular weight of less than 25 kDa. The expected weight is 27.8 kDa.
Sortase-mediated Ligation of the GGGG-tagged sfGFP
The protein was cleaved with TEV protease to produce the N-terminal polyG tag. The cleaved protein was then used in different characterisation assays of different Sortase variants.
The measured absorbance and emission spectra indicated that TAMRA and superfolder green fluorescence protein (sfGFP) are a possible FRET-pair. The sfGFP has an N-terminal polyglycine sequence and can therefore be linked to TAMRA with the sorting motif, in the same way as mCherry was connected. However, the small overlap between the extinction spectra of sfGFP and TAMRA could solve the previous “simultaneous excitation” problem we observed for the mCherry-TAMRA FRET-pair. Because of the lower excitation maximum of sfGFP compared to TAMRA, sfGFP was chosen as donor and TAMRA as acceptor. sfGFP was excited at 465 nm to minimize the unnecessary leak excitation of sfGFP.


Figure 2 : Design of a FRET-pair of 5-TAMRA-LPETG (TAMRA) and GGGG-sfGFP (sfGFP). In this configuration sfGFP acts as donor and TAMRA as acceptor. When the two fluorophores are not linked only sfGFP is being excited. After sortase-mediated ligation of the two substrates, TAMRA is the fluorophore being excited via FRET and the emission of TAMRA intensifies. Meanwhile, the emission of sfGFP decreases.
The transfer of energy from sfGFP to TAMRA can be seen by the decrease in emission of sfGFP and increase in emission from TAMRA. Compared to TAMRA as an acceptor, the sfGFP bleaches significantly less and is consequently more suitable as a donor for FRET. Furthermore, the afore mentioned problem of simultaneous donor and acceptor excitation seems to be solved. It seems that we have found a FRET-pair with superior properties.


Figure 3 : Spectrum of TAMRA and sfGFP, with Sortase A7M, over the course of 25 min in 5 min intervals. Depicted are the emission wavelengths against the RFU. The sortase-mediated ligation results in a decline of both emission peaks.
Due to the collected data of both FRET-pairs we decided to use the TAMRA-LPETG and GGGG-sfGFP FRET-pair for further characterization of our Sortase A variants. Two reasons justify this decision:
- TAMRA bleaches stronger than sfGFP when excited with a laser.
- The spectral overlap between TAMRA and mCherry disturbs “clean” energy transfer, thus the FRET-effect would be less visible and could not be used for analysis of the sortase-mediated reaction.
For recording of sortase reaction parameters we recommend using the FRET-pair sfGFP-TAMRA. As this pair of fluorophores proved to have near perfectly aligned spectra and since the bleaching effect is visibly lower on sfGFP than on TAMRA, we chose to use this FRET-pair in most of our following assay. Nevertheless, we do not rule out the use of TAMRA-mCherry as a FRET-pair since we used it in several FRET-assays as well.
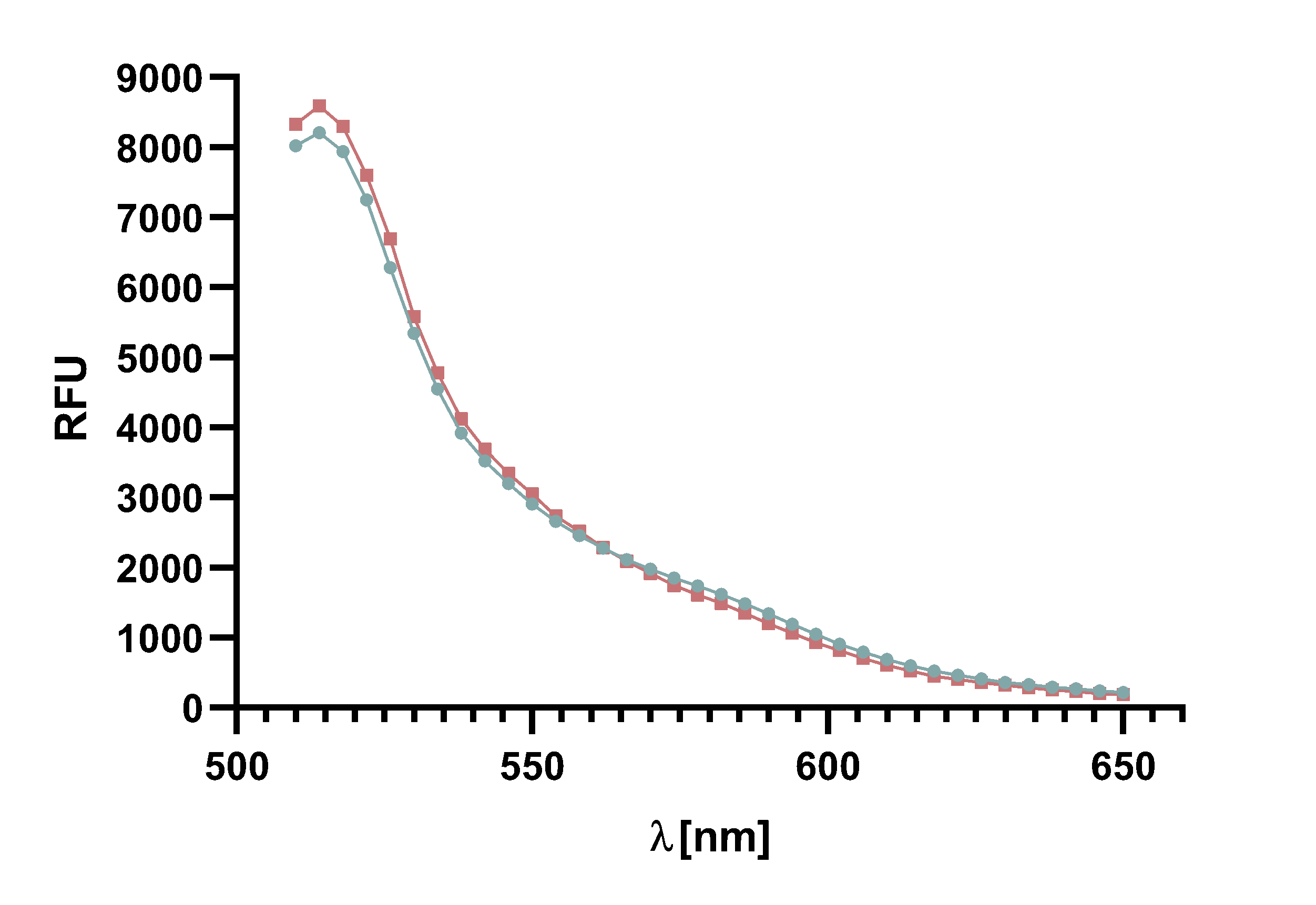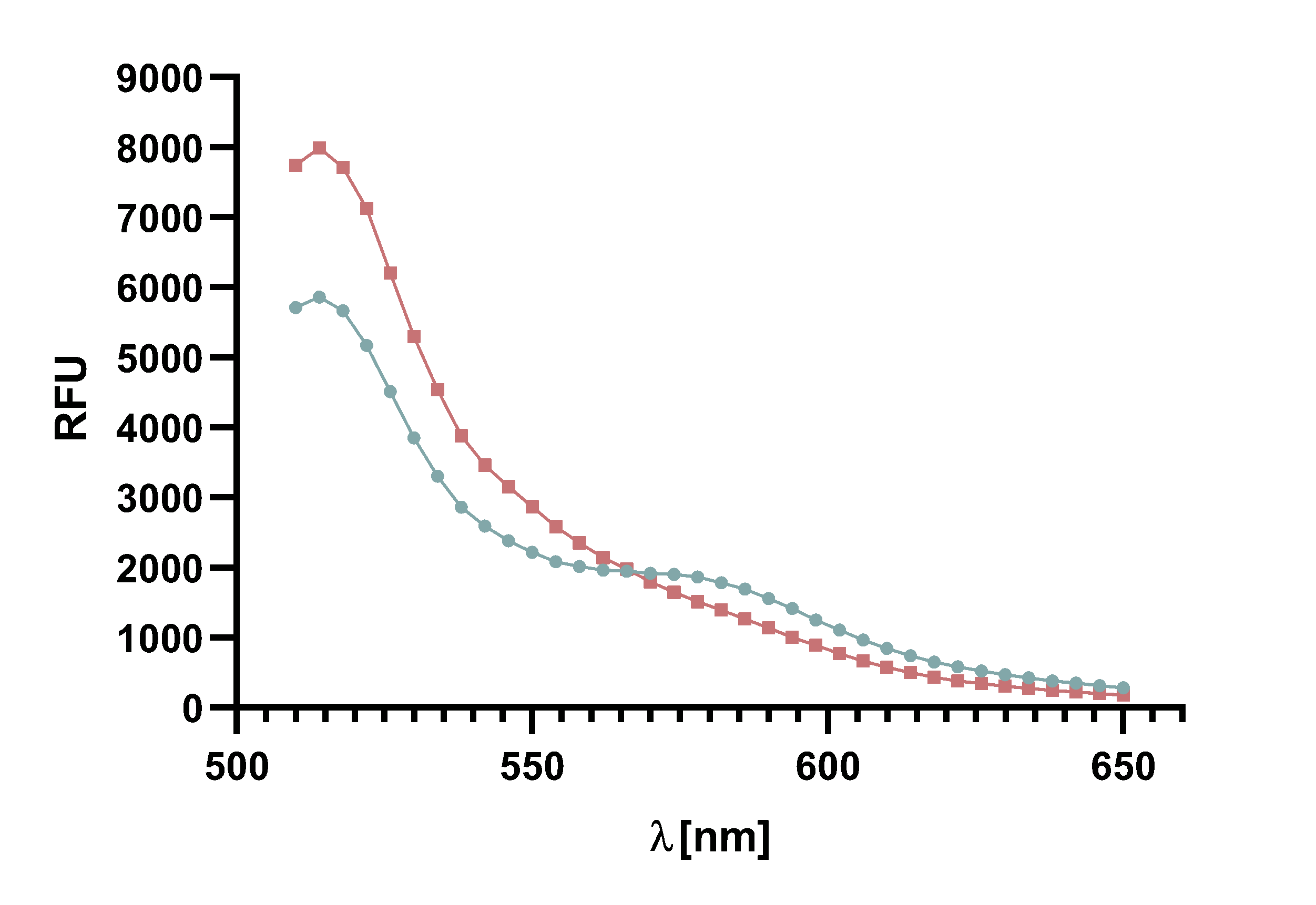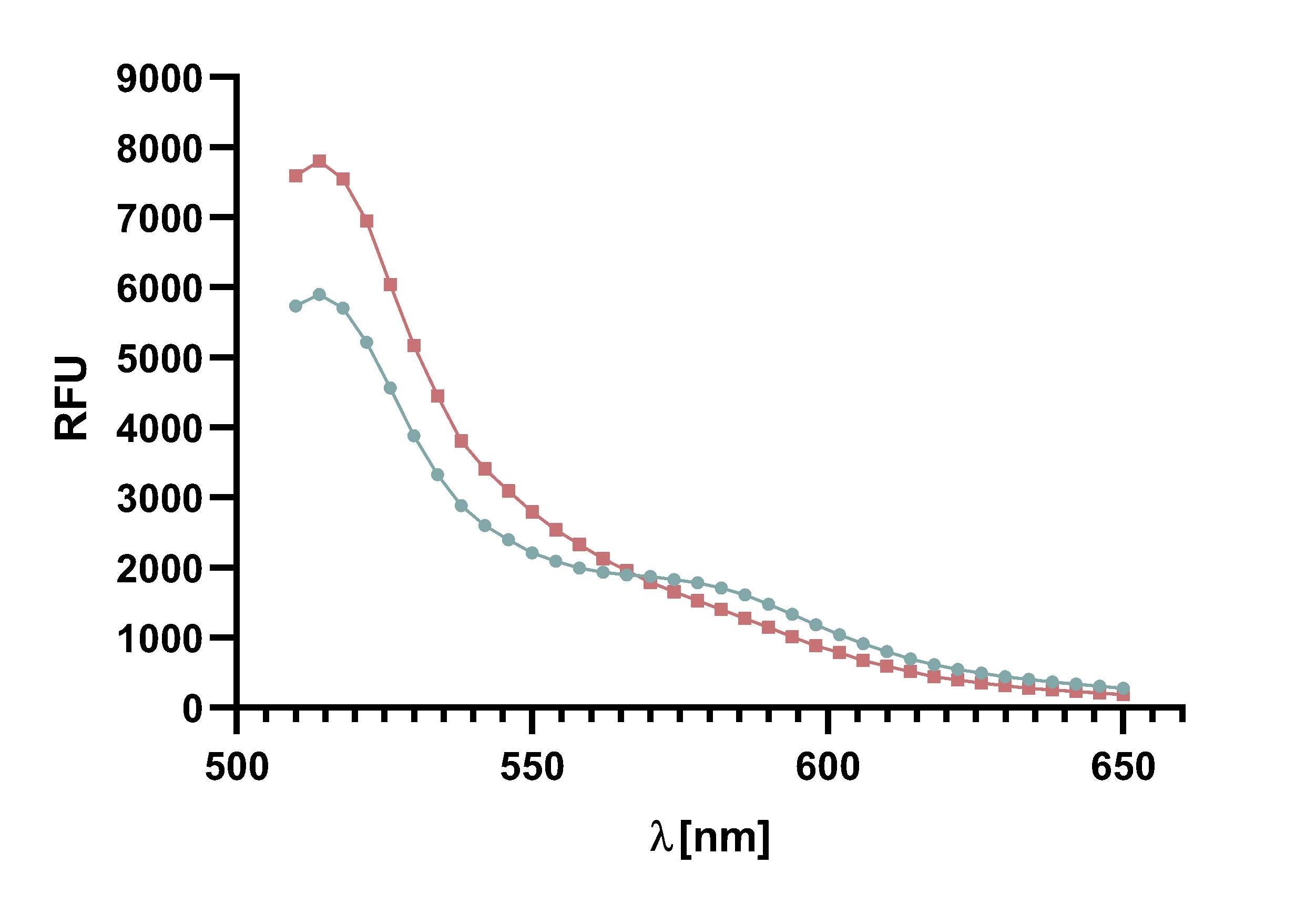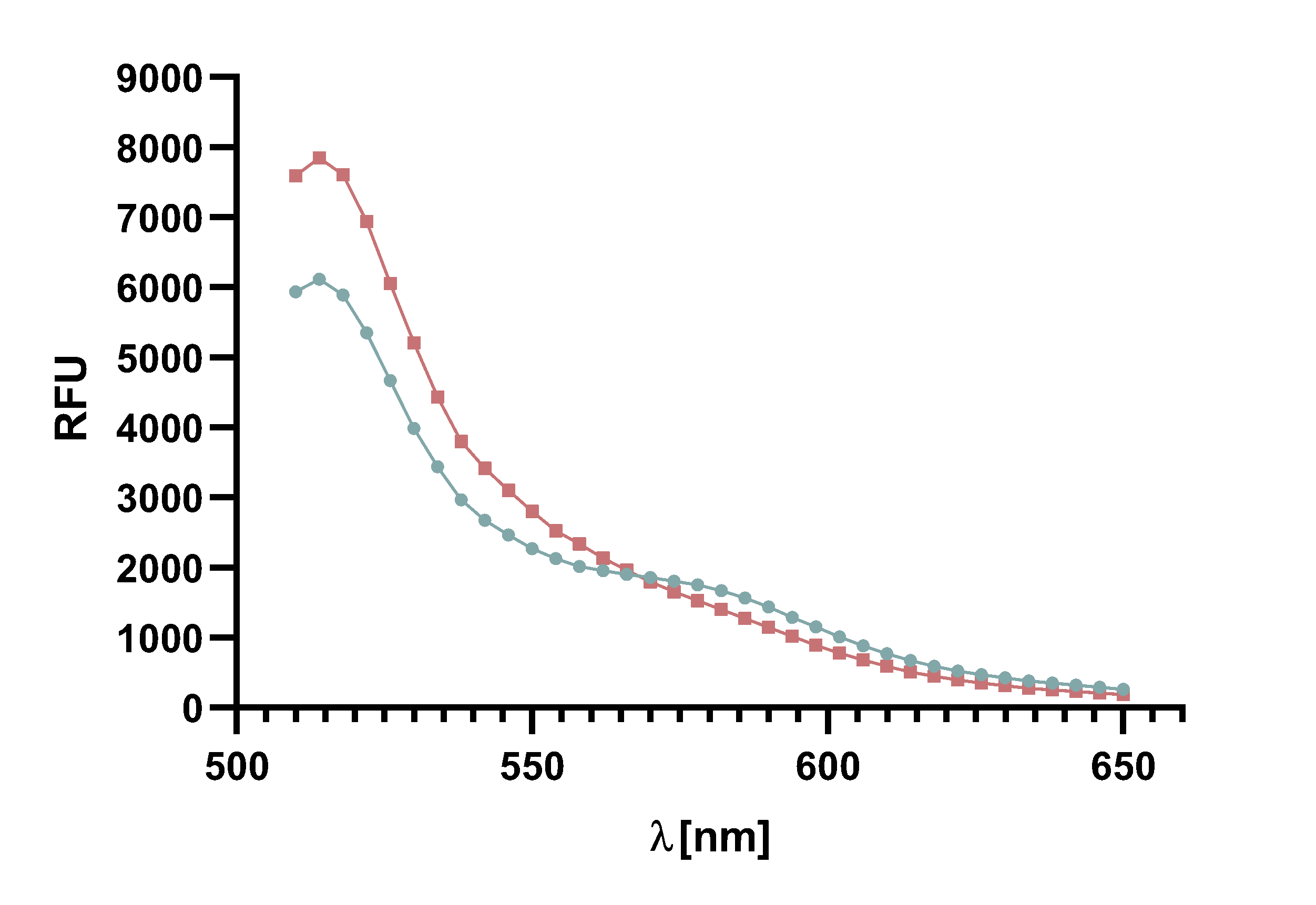
Figure 4 : Animation of Sortase A7M enzyme kinetics over the course of 3 h. The reaction speed increases radically in the beginning moving from RFU 8000 to RFU 6000 at λ = 550 nm where a plateau is reached (blue). The negative control (orange) is also reduced in its RFU due to bleaching. Nevertheless, a peak at λ = 580 nm arises already after short reaction time. This peak indicates the successful Fluorescence Resonance Energy Transfer.
VLP modification
The cleaved protein was further used for modification of assembled virus-like particles.
In order to demonstrate the integrity of our modified VLPs we used capsids from the same sample for DLS and electron microscopy which confirms the presence of intact VLPs. The size distribution shows that they still pose a monodisperse species, even though their hydrodynamic diameter is increased compared to unmodified VLPs or capsids containing only CP.

Figure 5: Dynamic light scattering analysis. Hydrodynamic diameters of different P22-VLP species.
We also used ultracentrifugation over a sucrose cushion to separate freshly modified VLPs from monomeric capsid proteins, Sortase A5M, and sfGFP. After ultracentrifugation, a green fluorescent sediment was clearly visible (Fig. 6). This is a strong indication that sortase has attached sfGFP to the VLP exterior, as only assembled VLPs accumulate in the sediment. [8] We then prepared the ultracentrifugation sediment for transmission electron microscopy. Encouragingly, we observed numerous visually intact VLPs.

Figure 6: Sediment containing P22-VLPs modified with sfGFP using SortaseA5M. Sediment was imaged in transmission electron microscope.
References
- ↑ W. Earnshaw, S. Casjens, S. C. Harrison, Assembly of the head of bacteriophage P22: X-ray diffraction from heads, proheads and related structures J. Mol. Biol. 1976, 104, 387. [1]
- ↑ W. Jiang, Z. Li, Z. Zhang, M. L. Baker, P. E. Prevelige, W. Chiu, Coat protein fold and maturation transition of bacteriophage P22 seen at subnanometer resolutions, Nat. Struct. Biol. 2003, 10, 131. [2]
- ↑ Dustin P. Patterson, Benjamin Schwarz, Ryan S. Waters, Tomas Gedeon, and Trevor Douglas, Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-Like Particle ,ACS Chemical Biology 2014 9 (2), 359-365 [3]
- ↑ Proft, T. (2010) Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation [4]
- ↑ Mao, H., Hart, S. A., Schink, A., and Pollok, B. A. (2004) Sortase-mediated protein ligation: a new method for protein engineering [5]
- ↑ Jean-Denis Pédelacq, Stéphanie Cabantous, Timothy Tran, Thomas C Terwilliger, Geoffrey S Waldo, Engineering and characterization of a superfolder green fluorescent protein, Nature Biotechnology volume24,pages79–88 (2006) [6]
- ↑ FPbase: Superfolder GFP, last visited: 10.6.2019 [7]
Sequence and Features
- 10COMPATIBLE WITH RFC[10]
- 12COMPATIBLE WITH RFC[12]
- 21INCOMPATIBLE WITH RFC[21]Illegal BamHI site found at 840
- 23COMPATIBLE WITH RFC[23]
- 25COMPATIBLE WITH RFC[25]
- 1000INCOMPATIBLE WITH RFC[1000]Illegal SapI.rc site found at 138